 "
"
Team:Bielefeld-Germany/Project/Protocols
From 2010.igem.org

Lab protocols
Preparation of electrocompetent E. coli cells
modified from [http://openwetware.org/wiki/Quint_Lab:electrocompetent_cells_e.coli Quint Lab]
Materials:
- 10 mL LB-Medium (1 % Bacto Trypton; 0.5 % Yeast Extract; 0.5 % NaCl)
- 2 L cooled bidest H2O
- 200 mL cooled, sterile-filtered 10 % glycerol
- Box with ice-water for 2-litre-flask
- 4 pre-cooled 250 mL (or 2x500 mL) bins for centrifugation
- 2 pre-cooled 50 mL Falcons
- Centrifuge, pre-cooled to 2 °C (max. 4 °C)
Protocol
- Inoculate 2x10 mL LB with bacterial stock; incubate over night at 37 °C and 200 rpm
- Inoculate 2x250 mL LB in 1-litre-flask with OD600=0.1 at 37 °C or at 19°C over night
- Incubate until OD600 0.4-0.6 (~5 h)
- The following steps are done at 2-4 °C (best in a cold room)
- Cool 1L-culture 10-15 minutes in ice water (shake sometimes)
- Divide culture into 2x5 cooled 50 mL Falcon-tubes for centrifugation
- Centrifuge 10 min at 4°C with 4000 rcf
- Discard supernatant
- Resuspend pellet in 5 mL bidest. H2O
- Add bidest. H2O up to 50 mL
- Centrifuge 10 min at 4°C with 4000 rcf
- Discard supernatant immediately
- Resuspend pellet in residual supernatant
- Add bidest H2O up to 50 mL
- Centrifuge 10 min at 4°C with 4000 rcf
- Discard supernatant immediately
- Resuspend pellet in residual supernatant
- Transfer suspension in 2x50 mL Falcons
- Add 10% glycerol up to 50 mL
- Centrifuge 5 min at 4°C with 4000 rcf
- Discard supernatant
- Estimate volume of the pellet; fill up with equal volume of 10% glycerol
- Resuspend pellet on ice; ‘’’don´t vortex!!’’’ (just shake cautiously)
- Divide cells into 150 µL aliquots (use 1.5 mL Eppis)
- Freeze in liquid N2 or dry-ice
- Store at -80°C
Preparation of heat shock competent E.coli cells
- Inoculate 2x5 mL LB with bacterial stock; incubate over night at 37°C and 200 rpm
- Inoculate 2x250 mL SOB in 1-litre-flasks with OD600=0.1
- Incubate at 19°C until OD600=0.4-0.6 (20h)
- The following steps are done at 2-4°C (best in a cold room)
- Cool 1 L of culture 10-15 minutes in ice water (shake sometimes)
- Divide culture into 2x5 cooled 50 mL Falcon-tubes for centrifugation
- Centrifuge 10 min at 4°C with 4000 rcf
- Discard supernatant
- Resuspend cells in 80 mL TB buffer
- Cool cells for 10 min on ice
- Centrifuge 10 min at 4°C with 2500 rcf
- Discard supernatant
- Resuspend cells in 20 mL TB buffer
- Add 1.5 mL Dimethyl sulfoxide (DMSO) for each 20 mL cells (DMSO concentration = 7%)
- Cool cells for 10 min on ice
- Divide cells into 150 µL Alicuots (use 1.5 mL Eppis)
- Freeze in liquid N2 or dry-ice
- Store at -80°C
Transformation via electroporation
- Thaw 50 µL competent E.coli cells on ice, dilute with icecold 50 µL glycerol (10 %) if necessary
- Add 0.5-5 µL plasmid to 50 µl electrocompetent cells
- Store cells on ice for 1 minute
- Electroporate at U = 2.5 kV, C = 25 µF, R = 200 Ώ
- Transfer transformation reaction to 450 µL SOC-Medium and shake 1 h at 37°C
- Centrifuge 2 min at 800 rpm and plate on selective LB-Medium
Heat shock transformation
- Thaw 150 µL competent E. coli cells on ice
- Add max. 10 µL DNA (the less the better your transformation works but at least about 50 ng vector)
- Incubate 30 min on ice
- Heatshock: 42 °C, 45 s (water bath because of quick heat transfer)
- 1 min on ice
- Add 1 mL prewarmed SOC-medium
- Incubate: 45 - 60 min, 37 °C
- Plate 100 µL
- Spin down the remaining cells (2 min, 5000 g), discard most of the supernatant, resuspend the cells in the remaining medium and plate them
Silver BioBrick Assembly
modified from [http://openwetware.org/wiki/Silver:_BB_Strategy Silver lab]:
This assembly method can be used for BioBricks which are bigger than 150 bp. The BioBrick should be at least 500 bp bigger or smaller than the backbone. The BioBrick, which complies with these conditions, is used as the insert and is assembled into the prefix or suffix of the other used BioBrick, called vector. So you have to differentiate between a prefix and a suffix insertion.


Suffix Insertion
- Digestion of insert: at least 700 ng DNA / 10 µL volume, 1 µL 10x Tango buffer, 0.5 µL XbaI, 1 µL PstI. Digest for 2 h at 37 °C, afterwards inactivation for 20 min at 80 °C. Clean up the insert via gel electrophoresis. When cutting the insert out of the gel try to avoid staining or exposure to ultraviolet light of the insert.
- Digestion of vector about 700 ng DNA / 10 µL volume, 1 µL 10x orange buffer, 0.5 µL SpeI, 0.5 µL PstI. Digest for 2 h at 37 °C, afterwards inactivation for 20 min at 80 °C. Add 1 µL SAP (shrimp alcaline phosphatase) and 1.2 µL 10 x SAP buffer, incubate for 1 h at 37 °C. Clean up the vector with a PCR clean-up kit.
- Ligation: after digestion and clean-up: 50 - 200 ng of vector, 3 - 10 fold molar access of insert, 20 µL ligation volume, 2 µL T4-Ligase-Buffer, 1 µL T4-Ligase. Incubate for 1 h at 37 °C, afterwards inactivation for 5 min at 70 °C. Then: store at -20 °C or transform.
Prefix Insertion
- Digestion of insert: at least 700 ng DNA / 10 µL volume, 1 µL 10x BamHI buffer, 0.5 µL EcoRI, 0.5 µL SpeI. Digest for 2 h at 37 °C, afterwards inactivation for 20 min at 80 °C. Clean up the insert via gel electrophoresis. When cutting the insert out of the gel try to avoid staining or exposure to ultraviolet light of the insert.
- Digestion of vector about 700 ng DNA / 10 µL volume, 1 µL 10 x Tango buffer, 0.5 µL EcoRI, 0.5 µL XbaI. Digest for 2h at 37 °C, afterwards inactivation for 20 min at 80 °C. Add 1 µL SAP (shrimp alcaline phosphatase) and 1.2 µL 10 x SAP buffer, incubate for 1 h at 37 °C. Clean up the vector with a PCR clean-up kit.
- Ligation: after digestion and clean-up: 50 - 200 ng of vector, 3 - 10 fold molar access of insert, 20 µL ligation volume, 2 µL T4-Ligase-Buffer, 1 µL T4-Ligase. Incubate for 1 h at 37 °C, afterwards inactivation for 5 min at 70 °C. Then: store at -20 °C or transform.
Variations
- A digestion over night is possible. If you digest over night use only 0.1 µL restriction enzyme.
- It is also possible to use PCR product as insert. Digest after PCR with corresponding restriction enzymes and clean up with PCR clean-up kit. This could lead to higher yields of insert DNA because a lot of DNA gets lost during the gel electrophoresis clean up.
- Sometimes some BioBricks are hard to assemble. Then you have to clean up the vector by gel electrophoresis as well.
3A assembly
Modified from [http://ginkgobioworks.com/support/BioBrick_Assembly_Manual.pdf BioBrick Assembly Manual by Ginkgo BioWorks]
Digestion
- Thaw DNA from upstream and downstream part and the destination plasmid on ice.
- Destination plasmid has to carry the ccdB gene <partinfo>P1010</partinfo> as insert and has to have a different antibiotic resistance than the plasmids carrying the upstream and downstream parts
- DNA has to be cleaned (by MiniPrep or after a PCR)
- 500 ng DNA / digestion mix for upstream and downstream part, 150 ng DNA / digestion mix for destination plasmid (total volume of mix 10 µL, dilute with ddH20 if necessary)
- Add 1 µL of buffer and restriction enzymes as shown in the following table:
| Upstream part | Downstream part | Destination plasmid | |
|---|---|---|---|
| enzyme 1 | 1 µL EcoRI | 0.5 µL XbaI | 0.5 µL EcoRI |
| enzyme 2 | 1 µL SpeI | 1 µL PstI | 0.5 µL PstI |
| buffer | BamHI | Tango | Orange |
- Incubation of the digestion mixes for 2 h at 37 °C, afterwards heat inactivation for 20 min at 80 °C
- Continue with ligation or freeze the mixes
Ligation
- Ligation mix:
- 2 µL ddH2O
- 5 µL of every digestion mix (so 15 µL in total)
- 2 µL T4-DNA-ligase buffer (thaw on ice!)
- 1 µL T4-DNA-ligase
- Incubate 1 h at 37 °C, afterwards heat inactivation for 5 min at 70 °C
- Freeze ligation mix or continue with transformation (heatshock or electroporation)
Phusion PCR
- Perform a Phusion PCR for amplifying BioBricks or gaining new BioBricks
- For one PCR-Reaction mix:
- 10 µL 5x HF-Buffer
- 4 µL dNTPs (2.5 mM each)
- 1.5 µL DMSO
- 31.5 µL H2O
- 1 µL Primer Mix
- 1 µL Template
- 1 µL Phusion DNA-Polymerase
- PCR program:
- Start: 30 sec, 98 °C
- 35 cycles of:
- 10 sec, 98 °C
- 30 sec, 58-66 °C (depending on primers)
- 15-30 sec / 1 kb template, 72 °C
- 10 min, 72 °C
- 4 °C until further processing
- Perform a gel electrophoresis:
- Check the fragment size
- Depending whether there are fragments with wrong size perform a clean up via gel electrophoresis or just with a PCR clean-up kit
Colony PCR
- Pick one colony with a sterile tip and elute it in 100 µL ddH20 or medium
- Store the colony in 4°C while colony PCR is running
- One reaction mix contains:
- 2.5 µL 10x buffer
- 0.75 µL MgCl2
- 1 µL dNTPs
- 0.5 µL primer mix (prefix/suffix primers or sequencing primers)
- 19.25 µL ddH2O
- 0.5 µL taq-polymerase
- 0.5 µL template
- PCR program:
- Start: 8 min, 98 °C
- 30 cycles of:
- 30 s, 98 °C
- 30 s, 60 °C
- 30 s / 1 kb template, 72 °C
- Finish: 5 min, 72 °C
- Gel electrophoresis: check the fragment size
- Plate the correct colony
Construction of a plasmid with R6K origin of replication (ori)

Cloning of R6K origin into plasmid pSB1C3
- Perform a restriction digest of pSB1C3 and pSB1A2::<partinfo>J61001</partinfo> with EcoRI and PstI
- Dephosphorylate restricted pSB1C3
- Separate BBa_J61001 and pSB1A2 backbone by agarose gel electrophoresis and extract the ~400 bp fragment (BBa_J61001)
- Purify restricted fragments with gel extraction kit
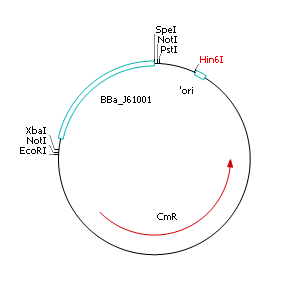
- Ligate restricted and purified pSB1C3 and BBa_J61001
- Transform ligated product into heatshock competent E. coli TOP 10 cells
- Streak out on selective LB agar plates containing chloramphenicol (10 µg µL-1)
- Incubate overnight at 37°C

Removal of pMB1/ColE1 origin of replication
- Streak out single colonies on selective LB agar plates containing chloramphenicol (10 µg µL-1)
- Incubate overnight at 37°C
- Isolate plasmid DNA
- Perform a restriction digest of pSB1C3::BBa_J61001 with Hin6I
- Separate fragments by agarose gel electrophoresis
- Extract and purify largest fragment (1758 bp) with gel extraction kit
- Religate extracted fragment
- Transform ligation product into heatshock competent E. coli EC100D cells
Comment: The resulting plasmid contains 32 remaining basepairs of the pMB1/ColE1 origin. This fragment does NOT enable replication in E. coli pir- strains.
Determination of minimal inhibitory concentration (MIC) of kanamycin
- Transform the plasmid K389212 (native virA) in bacteria which are already containing K389101 (kanamycin resistance read out) using electroporation.
- Plate 100 µL of the transformed bacteria in different steps of dilution (10-1 to 10-6) on LB-Agar with ampicillin (100 µg mL-1) and chloramphenicol (10 µg mL-1).
- Incubate the plates upside down at 37 °C for 16 – 20 h.
- Compare the plates of different dilution steps and select those, showing a high but countable number of colonies (aprox. 100 – 500).
In the following steps the colonies are transferred to LB-Agar with rising concentrations of kanamycin either in the presence of acetosyringone or without any inductor.
- Transfer the colonies of the selected plates to LB-Agars described below using replica plating. Take care to operate from lowest up to highest concentration of kanamycin. You must also use a fresh and sterile stamp every time you start a new sequence of plating.
- Composition of the LB-Agars (plating sequence from top to bottom)
- A) Uninduced status (no acetosyringone added)
| Positive control | Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (0 µM) | Kanamycin (0 µg mL-1) |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (0 µM) | Kanamycin (10 µg mL-1) | |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (0 µM) | Kanamycin (25 µg mL-1) | |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (0 µM) | Kanamycin (50 µg mL-1) | |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (0 µM) | Kanamycin (100 µg mL-1) | |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (0 µM) | Kanamycin (200 µg mL-1) |
- B) Induced status (200 µM acetosyringone)
| Positive Control | Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (200 µM) | Kanamycin (0 µg mL-1) |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (200 µM) | Kanamycin (10 µg mL-1) | |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (200 µM) | Kanamycin (25 µg mL-1) | |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (200 µM) | Kanamycin (50 µg mL-1) | |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (200 µM) | Kanamycin (100 µg mL-1) | |
| Ampicillin (100 µg mL-1) | Chloramphenicol (10 µg mL-1) | Acetosyringone (200 µM) | Kanamycin (200 µg mL-1) |
- Incubate the plates upside down at 37 °C for 16 – 20 h.
- Count the number of colonies on each plate.
- Calculate the ration of colonies grown on each plate, compared to the number of colonies on the first plate of each sequence (positive control).
Directed mutagenesis – Error-prone PCR and screening system
1. Error-prone PCR Step
Mix:
- 10 µL 100 mM Tris-HCL, pH 8.3
- 2.5 µL 2M KCL
- 3.5 µL 200 mM MgCL2
- 2 µL 25 mM MnCL2 (ddd immediately before initiation of the PCR)
- 4 µL 25 mM dCTP
- 4 µL 25 mM dTTP
- 4 µL 5 mM dATP
- 4 µL 5 mM dGTP
- 0.5 µL 100 µM 5´primer (equals 50 pmol)
- 0.5 µL 100 µM 3´primer (equals 50 pmol)
- x µL Template K389212 – total amount of 1.5 pmol
- 5 µL 1 U/µL taq DNA polymerase
- Add MiliQ water to 100 µL total volume
- PCR program:
- Start: 8 min, 98 °C (melt)
- 24 cycles of:
- 30 s, 98 °C (melt)
- 30 s, 60 °C (anneal)
- 30 s / 1 kb template, 72 °C (extension)
- Final extension: 5 min, 72 °C
- End: Keep at 4 °C
2. Cloning of virA variants
Clone the randomly mutated virA sequences (2.5 kB) under the control of the promoter <partinfo>J23110</partinfo> and the RBS <partinfo>B0034</partinfo> in the <partinfo>pSB1AT3</partinfo> backbone.
(See above for protocol of Prefix- / Suffix-Insertion)
3. Primary selection of novel virA variants
Transform the libraries of virA variants into electro-competent E. coli cells, in which <partinfo>K389011</partinfo> in a plasmid with R6K ori was already transformed and thereby the read out system via kanamycin resistance is already present.
(See above for protocol of transformation)
Plate the transformed bacteria on selective LB-Agar. When producing the plates add the following mixture of substances to the hand-warm LB-Agar:
- 1 mL ampicillin stock solution (final concentration 100 µg mL-1)
- 286 µL chloramphenicol stock solution (final concentration 10 µg mL-1)
- These antibiotica are used to ensure stability of both plasmids
- 50 mL substance-mix – including capsaicin, dopamine, homovanillic acid and 3-O methyldopamine (all final concentration of 200 µM)
- Any bacteria with a virA variation that is activated by at least one of these novel inductors will express the kanamycin resistance
- 2 mL Kanamycin stock solution (final: 100 µg mL-1)
- Compareable high kanamycin concentration to allow only these bacteria to grow which show a strong expression o the kanamycin resistance. (This optimal concentration was previously determined in MIC analysis.)
Incubate plates upside down at 37 °C for 16 – 20 h. The grown colonies will include a virA variant that is either constitutively active or had been induced by at least one of the tested substances.
4. Quantitative analysis of virA variants after induction with novel substances
To quantify the induction profile of the selected virA variants it is appropriate to change the read out system from kanamycin resistence to luciferase expression. This complex and time consuming task can easily be achieved without any cloning step, when using the advantage of our two plasmid system:
- Pick a colony of interest and plate it on a fresh LB-Agar, containing Amp (100 µg mL-1) and Cm (10 µg mL-1)
- Incubate upside down at 37 °C for 16 – 20 h.
- Isolate plasmids from the bacterial lawn with a miniprep.
- In this step two plasmids are isolated – One includes the virA variants in a normal <partinfo>pSB1AT3</partinfo> backbone. The second type with the kanamycin read out has the special R6K ori, which can only amplify in certain E. coli strains.
- Transform the isolated plasmids to a strain which is not capable to amplify the R6K ori (e.g. TOP10)
- Plate the transformants on LB-Agar with ampicillin (100 µg mL-1) and incubate upside down at 37 °C for 16 – 20 h.
- Isolate plasmids from the bacterial lawn with a miniprep.
- Here you are only harvesting the plasmid including the virA variant in <partinfo>pSB1AT3</partinfo>, since the other plasmid was not amplified in the grown E. coli.
- Transform the plasmid with the virA variant into competent cells, already including a second plasmid with a read out system luciferase or mRFP.
- Grow the transformants in shake flaks in LB-Media containing ampicillin (100 µg mL-1), chloramphenicol (10 µg mL-1) and 200 µM of one testing substance (e.g. capsaicin). As a control you should also grow the same bacteria without any inductor and with the native inductor acetosyringone.
- Take samples in certain intervals (e.g. 1 h) and measure the activity of the luciferase or mRFP.
Restriction analysis
- Digest BioBrick of interest: about 400 ng DNA / 10 µL volume, 1 µL 10x orange buffer, 0.5 µL NotI or PstI. Digest for 2 h at 37 °C. NotI is used to determine the length of the BioBrick and the plasmid backbone, PstI ist used to determine the length of the BioBrick in the plasmid backbone.
- Gel electrophoresis: add 2 µL loading buffer to every digestion mix, apply about 100 - 200 ng DNA / pocket in gel. Don't forget to apply the uncut BioBrick as well. A good agarose concentration for BioBricks between 0.2 and 3 kb is 1.5 %. The smaller your BioBrick of interest is the higher the agarose concentration should be and vice versa. The gel electrophoresis is made with TAE-buffer. Be sure that you melt your agarose gel in the same buffer you use for the electrophoresis later.
Cultivation for measuring mRFP and Luciferase expression
- Inoculate 10 mL LB containing desired Antibiotic with glyccerol stock
- Cultivate over night at 37 °C and 175 rpm
- Measure the OD600
- Prepare shake flasks with LB, antibiotic and inducer
- For Luciferase Measurement at least 10 mL starting volume
- For mRFP Measurement at least 20 mL starting volume
- Inoculate the main culture with a starting OD600 of 0.1
- Cultivate at 37 °C and 175 rpm
- Take a sample at least every hour and measure the OD600
- The sample handling depends on the readout strategy and is described in the corresponding measuring protocol ( mRFP or Luciferase)
Measuring of mRFP
- Take at least 500 µL sample for each measurement (200 µL is needed for one measurement) so you can perform a repeat determination
- Freeze samples at -80 °C for storage
- To measure the samples thaw at room temperature and fill 200 µL of each sample in one well of a black, flat bottom 96 well microtiter plate (perform at least a repeat determination)
- Measure the fluorescence in a platereader (we used a Tecan Infinite® m200 platereader) with following settings:
- 20 sec orbital shaking (1 mm amplitude with a frequency of 87.6 rpm)
- Measurement mode: Top
- Excitation: 584 nm
- Emission: 620 nm
- Number of reads: 25
- Manual gain: 150
- Integration time: 20 µs
Measuring of Luciferase
For the luciferase detection we used a [http://www.promega.com/tbs/tb281/tb281.pdf Promega Luciferase Assay System], containing a Cell Culture Lysis Reagent, Luciferase Assay Substrate and Luciferase Assay Buffer
Protocol:
- Prepare reaction tubes with 10 µL of high salt buffer (1M K2HPO4, 20mM EDTA, pH 7.8)
- Add 90 µL sample, mix and freeze at -80 °C
- For the measurement thaw by placing the tubes in room temperature water
- Add 300 µL of freshly prepared lysis mix ( 1X Cell Culture Lysis Reagent, 1.25 mg mL-1 lysozyme, 2.5 mg mL-1 BSA, add water for desired volume)
- Mix and incubate the cells for 10 minutes at room temperature
- Prepare the Luciferase Assay Reagent, by adding 10 mL of Luciferase Assay Buffer to the vial containing the Luciferase Assay Substrate
- Fill each well of a white, flat bottom 96 well microtiter plate with 20 µL of cell lysate
- For the detection of luciferase use a plate reading luminometer with injector for the Luciferase Assay Reagent and following settings (we used a Promega GloMax®-Multi Detection System with dual injector):
- Injection volume of Luciferase Assay Reagent: 100 µL
- Delay: 20 secs
- Integration: 3 secs
Chemicals, material etc.
Enzymes
| Enzyme | Producer |
|---|---|
| Pfu DNA-polymerase | Promega |
| Phusion DNA-polymerase | Finnzymes |
| Restriction enzymes | Fermentas |
| Shrimp alcaline phosphatase | Fermentas |
| T4-DNA-Ligase | Fermentas |
| taq DNA-polymerase | Bioline |
Kits
| Function | Name |
|---|---|
| Plasmid purification | Fermentas GeneJET™ Plasmid Miniprep Kit |
| PCR Cleanup | Macherey Nagel NucleoSpin® Extract II |
Inducer Stock Solutions
- Acetosyringone: 20 mM solved in 10 % (v/v) DMSO and 90 % (v/v) H2O
- Capsaicin: 20 mM solved in Methanol
- Dopamine: 20 mM solved in H2O
- Homovanillic acid: 20 mM solved in H2O
- 3-Methoxytyramine: 20 mM solved in H2O
TAE buffer
For 1 L of 50 x TAE buffer you need:
- 242.48 g Tris
- 41.02 g Sodiumacetate
- 18.612 g EDTA
- Adjust pH to 7.8
- Solve in dH2O
20 mL of the stock is diluted in 1 L dH2O for the gel electrophoresis.
DNA loading buffer
- 50 % (v/v) glycerol
- 1 mM EDTA
- 0.1 % (w/v) bromphenol blue
- Solve in ddH20
LB medium
For 1 L of LB medium you need:
- 10 g Trypton
- 5 g yeast extract
- 10 g NaCl
- 12 g Agar-Agar (for plates)
- Adjust pH to 7.0
Cell Culture Lysis Reagent
- 25 mM Tris-phosphate (pH 7.8)
- 2 mM DTT
- 2 mM 1,2-diaminocyclohexane-N,N,N´,N´-tetraacetic acid
- 10 % glycerol
- 1 % Triton® X-100
References
- Openwetware: Quint Lab: electrocompetent cells e.coli, [http://openwetware.org/wiki/Quint_Lab:electrocompetent_cells_e.coli http://openwetware.org/wiki/Quint_Lab:electrocompetent_cells_e.coli], October 24th 2010.
- Openwetware: Silver: BB Strategy, [http://openwetware.org/wiki/Silver:_BB_Strategy http://openwetware.org/wiki/Silver:_BB_Strategy], October 24th 2010.
- BioBrick Assembly Manual, Ginkgo BioWorks, [http://ginkgobioworks.com/support/BioBrick_Assembly_Manual.pdf http://ginkgobioworks.com/support/BioBrick_Assembly_Manual.pdf], October 24th 2010.
- Luciferase Assay System Technical Bulletin #TB281, Promega Cooperation, [http://www.promega.com/tbs/tb281/tb281.pdf http://www.promega.com/tbs/tb281/tb281.pdf], October 24th 2010.