 "
"
Team:USTC Software/html5
From 2010.igem.org
(New page: <html xmlns="http://www.w3.org/1999/xhtml" xml:lang="en" lang="en"> <head> <meta http-equiv="Content-Type" content="text/html; charset=UTF-8" /> <meta name="author" ...) |
|||
| Line 350: | Line 350: | ||
<!-- ContentBox content: page home --> | <!-- ContentBox content: page home --> | ||
<div id="pageHome" class="contentPage" style="visibility: visible;"> | <div id="pageHome" class="contentPage" style="visibility: visible;"> | ||
| - | </html> | + | </html><html> |
<img src="http://home.ustc.edu.cn/~soimort/igem_wiki/images/header.jpg" height=160 width=750 /> | <img src="http://home.ustc.edu.cn/~soimort/igem_wiki/images/header.jpg" height=160 width=750 /> | ||
Revision as of 02:55, 17 October 2010

- Overview
-
This is a paragraph
This is a paragraph
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Proin consectetur, ante non iaculis suscipit, massa tortor dictum massa, mattis iaculis massa odio sit amet ipsum. Integer posuere enim ac felis feugiat auctor. Ut urna dui, mollis hendrerit fermentum non, lacinia non enim. Vestibulum lacus risus, tempor vel egestas at, laoreet id tortor. Cras augue sapien, cursus in facilisis id, bibendum a enim. Curabitur semper ligula et ligula aliquet scelerisque. Nunc quis aliquet sem. Duis a rhoncus enim. Integer lacinia, mi.
- Features
-
Overall Project
The ultimate goal of USTC_Software 2010 team is to promote synthetic biology throughout the world. To attract more people who do not have biology background to be interested in this area, we plan to develop a modeling-and-simulation game specially designed for synthetic biology. Users are taught to learn the basic knowledge in the area via constructing their genetic circuits as input to our software and try to understand the system behavior as output. This is the basic functional module. We also plan to develop a rating system to grade users' design for given tasks, and a more friendly game interface that can fill the process of input with joy and ease. Though more functions are expected, we focus on the basic module, modeling-and-simulation, at the first stage of our project. In practice, many CAD (Computer-Aided Design) tools, such as TinkerCell and Synbioss, have been developed to model and simulate biological systems and give the system behaviors as guides. However, they all need users to provide details of the system network, such as the activation and repression of transcription and translation reactions in genetic regulatory network. It is so difficult for even professionals to construct a detailed network of a complex system depending only on their minds. In this sense, modeling through CAD tools will not reduce the work of modeling: users are actually required to model manually and input their model in details. However, since our software is developed for non-biological background users, it is unrealistic to expect them to model their design manually. To solve this problem, the USTC_Software 2010 team attempts to take synthetic biology modeling one step further by introducing new methods for automatic modeling of biological systems. Just as the word automation implies, users are only required to submit their assembling of parts, and the generation of biological model is automatically done by our program. Being the first-ever team trying to develop a synthetic biology automatic modeling tool, we focus on genetic regulatory network for the first year and develop our software tool, iGaME, which will assist the design of genetic function modules for biological systems in synthetic biology. We believe this will greatly relieve users from handling complex interactions of species in biological system. Many novel and revolutionary concepts are proposed during our development. The first is our Chain-Node model for complex structure with multiple chains bound together. It is necessary for automation since behaviors of species (such as how it react with other species) must be determined by their structures instead of their names: it is impossible to construct a universal name-based reaction database applying for different systems. The next is Template. A species with a certain structure is a template species, and a reaction occurring between several template species is a template reaction. The introduction of Template makes it possible to describe a group of reactions with same structure-determined reaction mechanism. Finally, based on our Chain-Node and Template concept, we propose an XML-based Standard Biological Part Automatic Modeling Database Language (MoDeL for abbreviation) to fully characterize Species and Reaction in templates with clear definitions of elements and attributes in XML fashion. It is a database language for next generation when automatic modeling is widely used throughout the world. To know more, users are suggested to read this One-Minute Introduction to have an intuitive idea of our modeling system. Though we have no enough time to achieve our ultimate goal completely, we have successfully developed a MoDeL-based, automatic modeling and simulation software. Our program falls into 3 major components. The first component is User Interface . Users could give their assembling of parts by drag-and-drop function and setup initial conditions as well. System behavior as output will also be shown there. The second component is database written in MoDeL , which is the kernel of our automatic modeling idea. The last component is our Core Program designed to support MoDeL language. It functions as a driver: completing system network based on users' input and data stored in our database to give dynamic analysis as output. - User Interface
Welcome to the world of creation and imagination!
As the world enters upon an age of human nature destructions, of resource starvation and the ignorance of ecological balance, the earth is more and more under the control of human beings, there is less room for the survival of animals, various kinds of microorganism are being killed and regenerated circulatory in laboratory.
However, there exsits a special kind of bacterial, who is too diligent to be willing to be controlled by human, they use their magic power to learn certain skills to being survived successfully. Reviewing their developmental history, their ancestors made great contribution to the world under successive human experiments. They show us the meaning of Biobrick, they grasp the ability to adapt themselves better to the outside world, they know how to realize their value furthest and greatly, they are the E.COLI!
Now let us begin the E.COLI magical mystery tour~~
Input
In order to give users a clear and definite goal on input. The whole input process is divided into two main steps: Compartment Construction and Plasmid Construction. In the first step, users only need to focus on the input of environmental condition, compartment, spatial structure of compartments, and the species contained in each compartment. In the second step, only plasmids are editable, and users can click on each of them to assemble biobricks into functional device. Of course, useful tips will be given along with your input process.
We here give a design of our user interface of input. It is under construction.
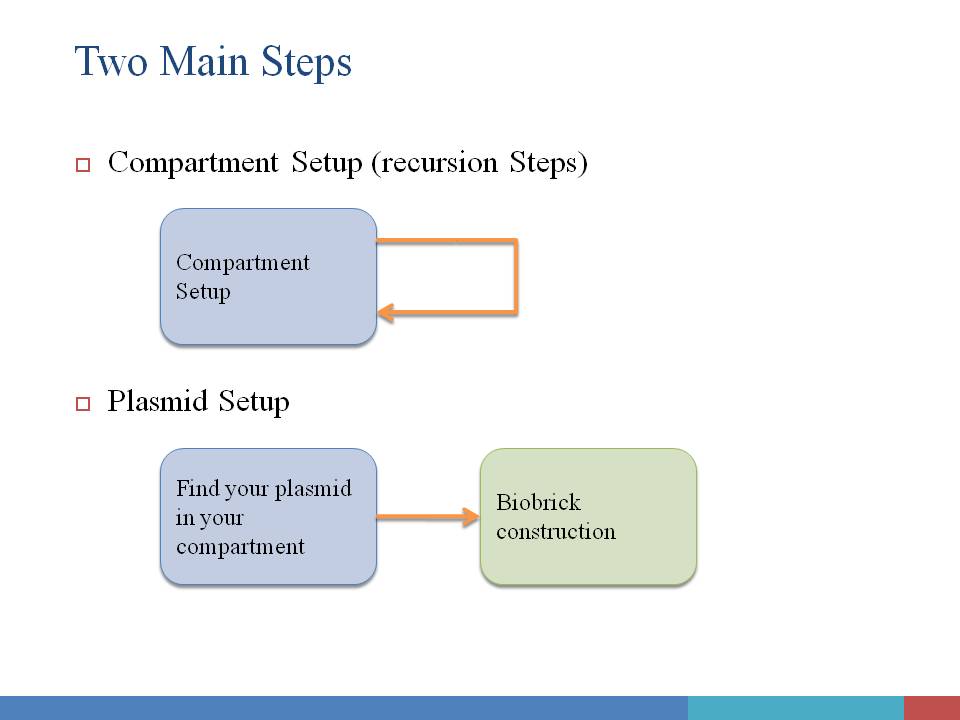

Step#1 Compartment Construction

Left: When you enter in our interface,the first step is to choose the environment of our system . A flask is like a container , all the reactions are happening in it . After that you may pick a appropriate condition for our lovely E.Coli . One E.Coli isn't enough for some specific problem , so you have to decide how many you will pick up . Small molecule is also available in our interface , you can decide their amount per second . They can be static or changing all the time . It's up to you !
Right: You can choose E.Coli and set their parameters one by one . Plasmid must be set up in this level . So you can smoothly enter into next level to make you own code of genes~~
OK!Now it's the time to give our system some more data , write down your favourite condition , it's good to be god , huh?~ Take it easy , next part is more attractive !
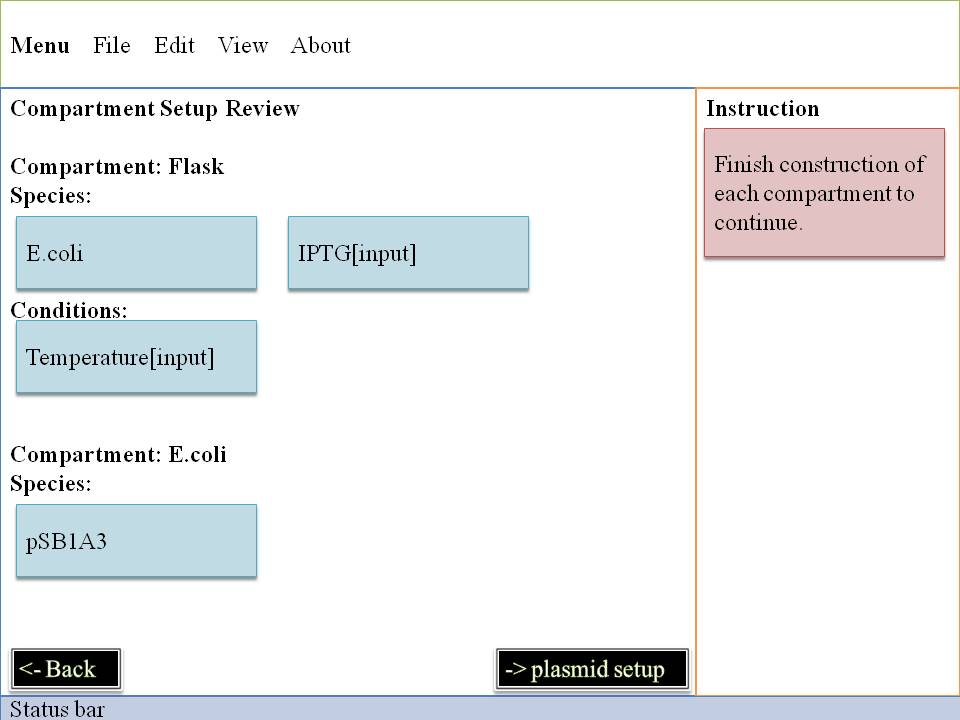
Step#2 Plasmid Construction

Here's the most interesting part! You can change the fundanmental construction of our cute Mr.ecoli! First you must locate on which E.coli you are interested in and then click it , second you may decide which plasmid to change .

Now we enter into the deepest level , we will control E.Coli with our own hand by dragging and dropping some biobricks .
Click - Drag - Drop , three simple motion can make a totally different Mr.ecoli . Think about that , thousands of E.Coli is waiting for you to rebuilt them , their willing eyes are looking at you , and expect you could help them make a difference . What are you wating for?! Just do it ! And E.Coli family will appreciate what you do~
OK~~Pay attention to what you pick up ! It's very important ! It can directly effect the way our system goes ~ So glare your eyes , use your mind to pick biobrick for our E.Coli . Do you want to see what you've created? Do you want to be god? Click on "simulation" ,and all your dreams will come true ! Show your imagination and talent to the microworld ! To your sincerely E.Coli~~
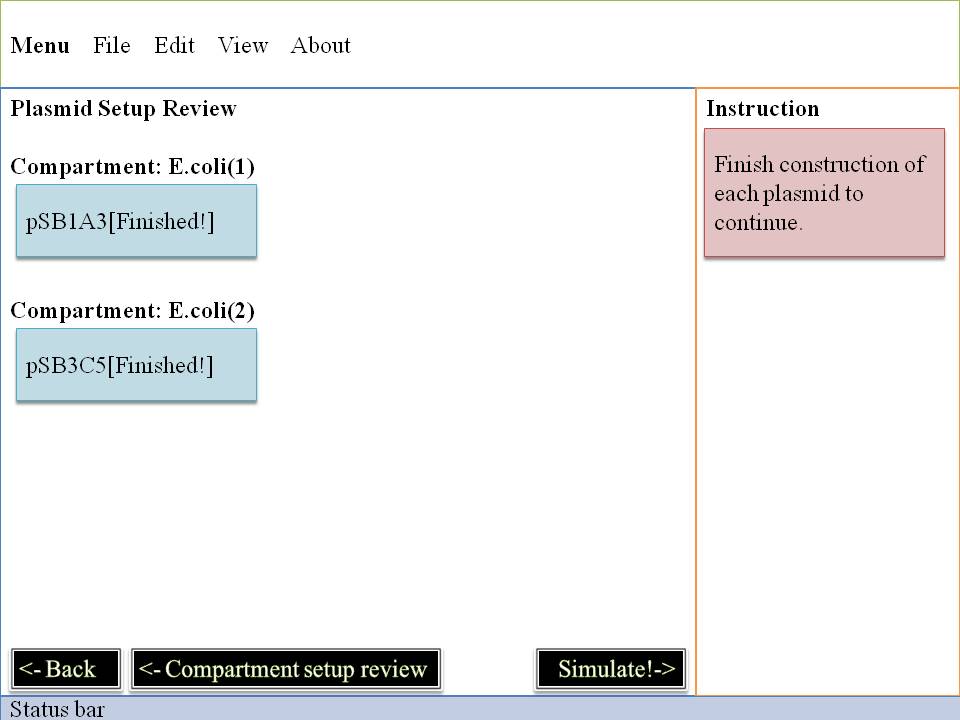
See Your Result!
Click Simulate button and see what will happen! Read more about our modeling idea & modeling algorithm!

Output
We use the powerful open source simulator for biochemical networks - COPASI, as the output interface of our software. It has many functions which will satisfy our needs in most situations. The tasks allowed includes steady-state analysis, time course, parameter scan, etc. The most frequently used function is to perform time course analysis. Users could obtain dynamic curves easily. What's more, it provides convenient way to modify parameters, species and reactions. Users could understand the model's network easily with friendly user interface.
- MoDeL
One-Minute Introduction
You are about to begin an exciting journey of the main ideas in MoDeL. For each picture, start from the green flag, follow the arrows, and finally you will reach the destination! Read on from the first step or click one of the three steps!
Chain-Node Model -> Template Modeling -> Automatic Modeling Database Language

Chain-Node Model
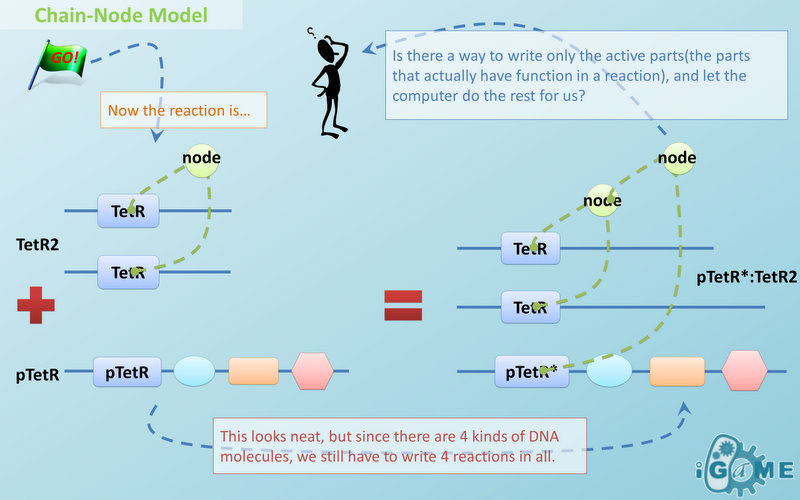
Template Modeling
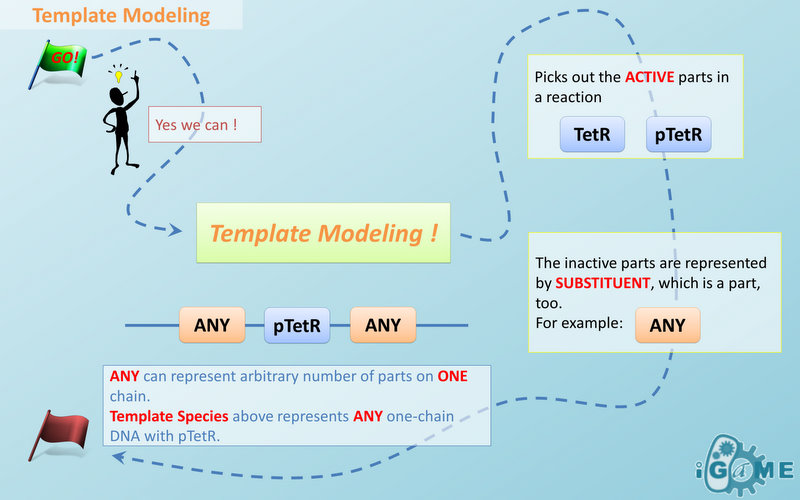
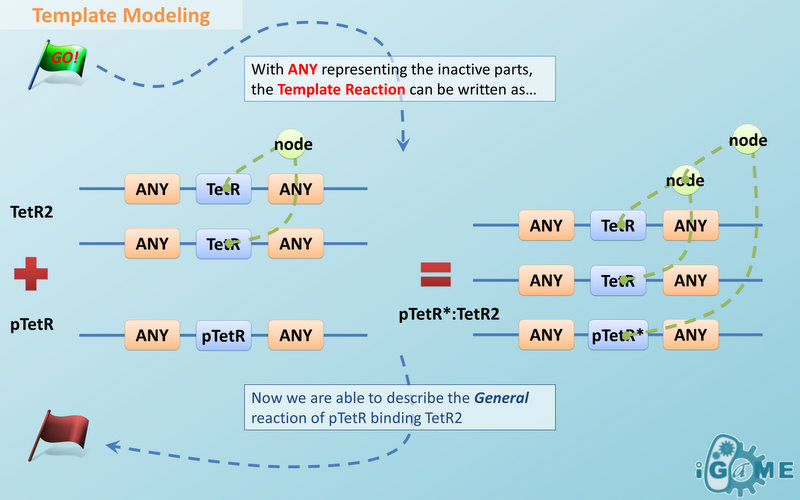
Automatic Modeling Database Language
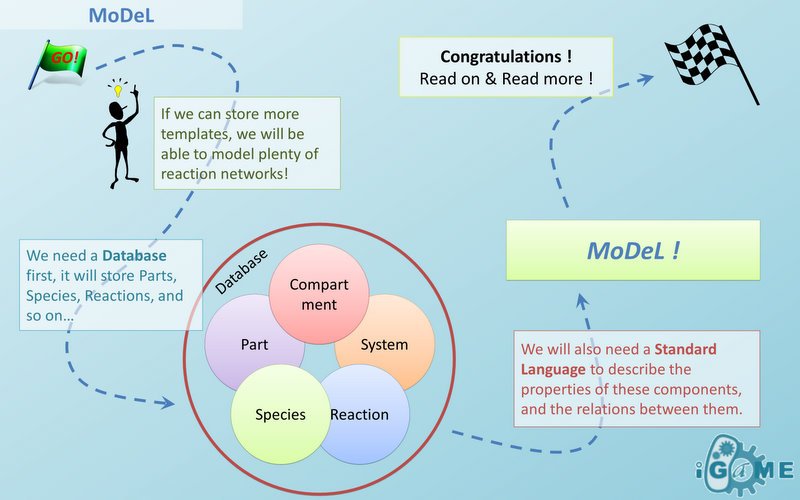

- Modeling
Introduction
We design and develop a software tool, iGame, which supports database written in MoDeL format, to perform task of automatic modeling. Actually, automatic modeling is a database-dependent network-searching-and-developing process from the initial conditions. Besides the database, users are required to provide initial environment condition, such as initial plasmids with biobrick parts. iGame will generate the network of the biological system in SBML standard. Please refer to User_Interface user interface to know more about input and output of iGame.Algorithms for automatic modeling will be shown in the following sections. An overview is as follow:
- Section Algorithm flow chart gives an overall description of the general framework of our algorithms;
- Section Special reactions and volume strategy tells details about the methods and strategies our program utilizes to handle with some special reactions;
- Section Chain Sorting and Weighting Scheme provides details of sorting as well as weighting scheme and their functions;
- Section Structural pattern match of template species provides details of the core algorithm of iGame: how to find matchings between template species defined in database and species existed in the bio-system;
- Section Generation of products based on reaction templates describes the mix-trim-split method to generate products.
Algorithm flow chart
Before stating our own algorithm, we will first make an analysis about the difference and difficulty brought by introducing a new language system. The general algorithm idea is common: find out all species and reactions between them. Ignoring the efficiency of program, the most obvious method is doing the following recursively until no more species is generated: - find reactions between species have been found and generate products as new species
Similar idea applies to our program. Some modifications must be made because in other software tools, reaction-searching is name-based. However, in our program, it is structure-based. Matching of species name is much easier than that of Chain-Node structure, which is featured as the most important part of our program. In addition, process of products generating is not as easy as just creating a new species with specific name, too. Their structures are required to be constructed under rules defined in MoDeL at the same time. The species matching and products generating modules are essential to implement our automatic modeling idea.
To complete the final systemn network from initial conditions, we break up the task to several parts:
- Setup initial conditions and environmental parameters read from the input file;
- Species produced are inserted into a list in the order of time. For each species, we search the database to find template species which could be matched to this species in structural pattern. If not found, then go for the next species;
- For each template species found matching, we continue to search template reactions containing this template species as a reactant or modifier or both. Only forward or reverse reaction is handled, never for both at the same time;
- For each template reaction found possible to occur, we search in the species list for possible species which could be instances of templates of other reactants and modifiers. If not found, then go for next reaction;
- For each possible combination of reactants and modifiers, if the reaction parameter conditions are satisfied for given constraints, we generate the structure of products based on the transfer table and structural templates of products. And then add this new reaction to the reaction list.
- Go to step 2 until the end of the species list;
In the following, we will focus on the step (2), (3) and (5). Our algorithms are totally different with that of other software tools since we design them for the purpose of proving the feasibility of MoDeL for synthetic biology. They are kind of complicated because the complexity of biological systems require so.
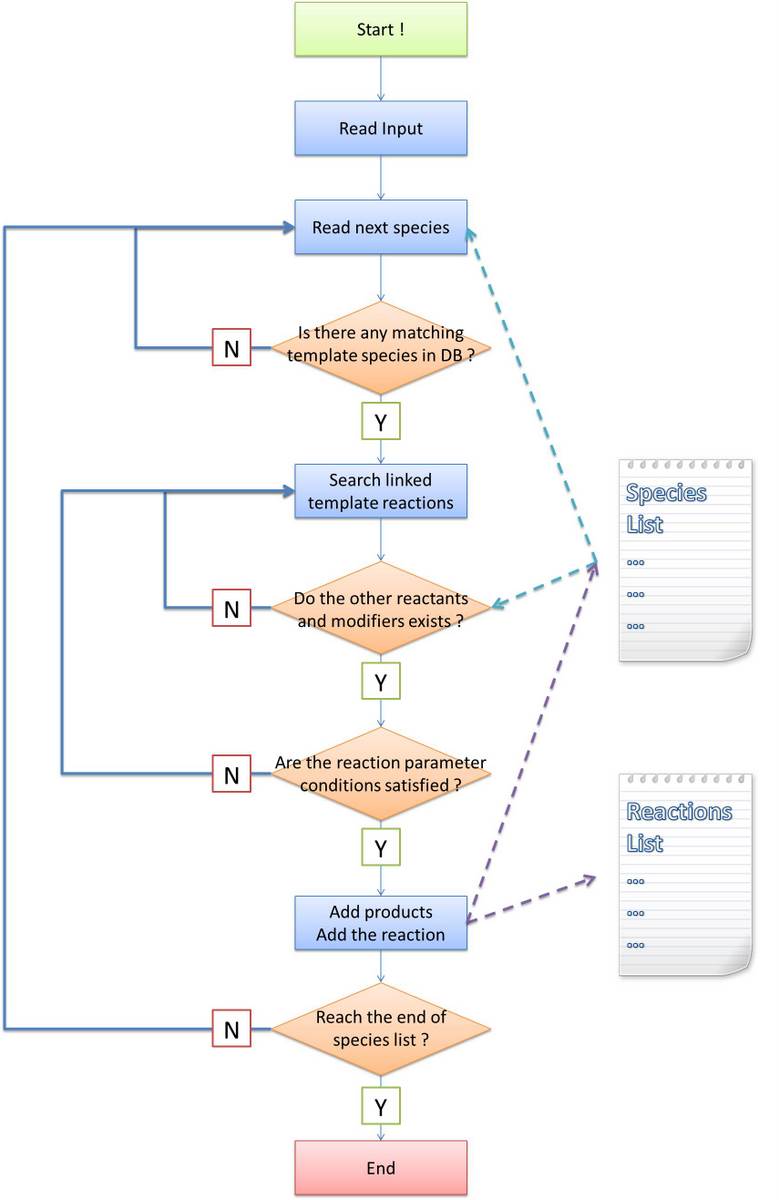 Figure 1: Algorithm Flow Chart
Figure 1: Algorithm Flow Chart
Special reactions and volume strategy
Transcription and translation reactions are not supported by MoDeL reaction definitions; they are handled specially in the core program. We use pseudo first-order reaction to model both transcription and translation reactions. The rate constant k is the value of forwardPromoterEfficiency/reversePromoterEfficiency for transcriptions and forwardRbsEfficiency/reverseRbsEfficiency for translations defined for each part in the database. It means number of mRNAs per DNA per second for transcriptions and number of proteins per mRNA per second for translations. A transcription will not end at a terminator with termination efficiency less than 100% (values are stored in attribute forwardTerminatorEfficiency/reverseTerminatorEfficiency): RNA polymerase will pass over and continue transcription. For translation reactions, the prerequisite condition is that the part next to RBS along the 5' to 3' direction must have non-zero attribute value forwardStartCodon/reverseStartCodon. Opposite to transcriptions, translation reactions would stop if one part with non-zero attribute value forwardStopCodon/reverseStopCodon is encountered.Volume strategy is a special method to solve problems introduced by diffusion reactions. Consider a transport reaction S1->S2 in which the species S1 is moved from the first compartment with volume V1, to the second with volume V2. The rate constant is k but the rate law is not k*[S1]*V1, because the concentration conversion is needed. The correct expression is N*k*[S1]*V1. The volume strategy just uses N*V1 instead of V1 as the volume of the first compartment. To have a general definition, the volume of a compartment is defined as V*N1*N2, where:
- V is the volume of the compartment itself;
- N1 is the number of compartments located in its outside compartment;
- N2 is the total number of its outside compartment
Chain Sorting and Weighting Scheme
Chains in our Chain-Node model should not have any order since there are no natural ways to sort them. One chain is not preferred to another, since in a complex of multiple chains, which are connected via binding interaction of binding bites on their own, all chains are equivalent. However, a chain-sorting program is a prerequisite for implementations of many other functions, such as comparison of two Chain-Node structures, or pattern matching of template species to those in the system.Sorting is always performed under some rules. Since there are no natural options, we design one to generate the unique order for a list of chains. The rule we developed is something like that for comparing two strings: compare the first part of two chains by their partReference and partType in alphabet order and if equal, then compare the next until there are differences or to the end of either chain. At this stage, besides partReference and partType, two parts are considered equal if both are in bound states or not. There are some exceptions for DNA molecule: since dsDNA has both forward and reverse directions, comparison between its forward and reverse chain is also needed.
If all chains are different in the first stage of sorting, we have already obtain the unique order of the chain list. However, it is not always so: equal chains in the above sorting may be equivalent or not. It is due to the assumption that two bound parts are taken as equal if both have same partReferenceand partType attribute values. Though the above sorting strategy takes binding positions into consideration, it ignores the binding details of those parts: the details are hidden in the nodes of all trees. Hence, similar to the chain sorting, we need to find another strategy to sort nodes as well as trees. Our weighting scheme will be first introduced below.
Assume the chains are sorted already. Each part will be assigned a weight based on its position in the chain list. The weight of the jth part on the ith chain is (i,j). For example, the first part on the first chain posses a weight (0,0) (in C style, counting number from zero). After assignment of parts' weight, we turn to weights of the nodes in trees. Since leaf nodes in trees are actually the representation of parts in bound state on chains, the weight of each leaf node could be inherited from the weight of its corresponding part. For parent node, its weight equals to the maximum of weights of all its children nodes (we define a weight (i, j) is greater than another (i',j') if i < i' or i = i' while j < j'). The recursive definition of nodes' weight of trees makes it possible to sort all children of each node, which is a prerequisite for generating Huffman codes (see below). And then the tree order in the forest could be uniquely determined by sorting the root node of each tree.
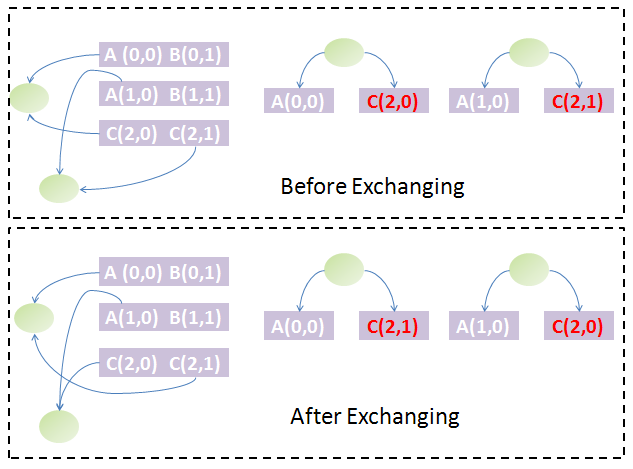 Exchange of the first and second chain. The tree structures are different before and after exchange: the two chains are not equivalent.
Exchange of the first and second chain. The tree structures are different before and after exchange: the two chains are not equivalent.Back to the question above, if two chains are equal in the first stage sorting, which should be arranged in front of the other? The answer could be obtained by exchanging them. The exchange will change the parts' weight on both chains and thus the weight of some nodes of the trees and subsequently the tree and forest structure. If the forest structure is not changed, the two chains are equivalent, which means that it makes no difference to arrange either chain in front of the other. If the forest structure has changed, we must find some ways to determine which tree structure is preferred and based on this to arrange them. We know each tree could be uniquely converted to a binary tree by taking the first child of each node as the left child and its siblings as its right child. The Huffman algorithm is used to generate the Huffman code of each node in the binary tree. Since the Huffman code of each node represents its path to the root node, we can restore the tree structure from the Huffman codes of all leaf nodes. Hence, the Huffman codes of leaf nodes could uniquely determine a tree structure. With help of Huffman codes, it is simple to compare the forest structure after and before exchange of both chains.
To go further, we must define the preference of each forest structure due to exchange of chains: compare each node of the first tree by their Huffman codes and weights in the same way as comparison of strings and if equal, compare the next until difference appears or to the end of either forest. If one is less than another, it has higher priority. With this definition, the order of those equal chains in the first-stage sorting is determined by the one which generates the forest strucuture with highest priority among all possibilities. By performing this algorithm for all groups of equal chains obtained from the first-stage sorting, we could find both the unique order of the chains and the groups of equivalent chains (exchange of the order fo several chains in the same equivalent group will not change the forest structure). The figure illustrates this idea graphically.
It is easy to compare two structures with help of chain sorting. We only need to compare first the chain list in order, chain by chain, and if equal, compare both associated forest structures. If two structures are same, the comparison must be equal since chains and trees are all in their unique order. If the comparison is equal, they must be the same structures since we could uniquely generate a Chain-Node structure from the chains as well as its associated forest structure.
Structural pattern match of template species
We assume that if a combination of species in the species list could be structurally matched to reactants and modifiers templates defined within a reaction, they are possible to react with each other in the way as the reaction template describes. Hence, it is important to find such matchings between species in the species list and template ones defined in the database. We will first give the basic idea and then some modifications.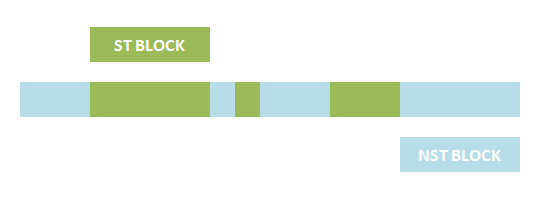 Blocks of ST and NST part on a template chain
Blocks of ST and NST part on a template chain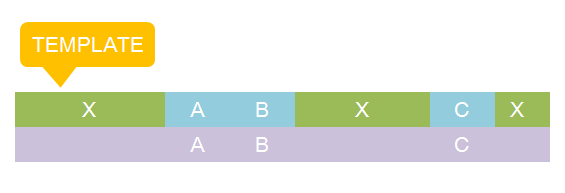 Combinations of NST block matchings form a region restriction for ST part blocks
Combinations of NST block matchings form a region restriction for ST part blocks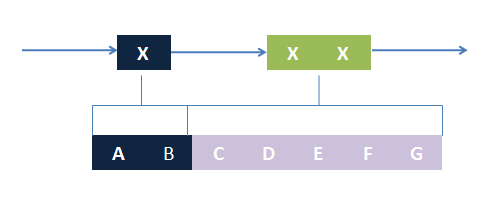 The template ST block contains 3 ANY parts. If the first ANY part is matched A and B, the task is reduced to match the next 2 ANY parts to C, D, E, F, and G. All matchings could be found by recursively performing this algorithm.
The template ST block contains 3 ANY parts. If the first ANY part is matched A and B, the task is reduced to match the next 2 ANY parts to C, D, E, F, and G. All matchings could be found by recursively performing this algorithm.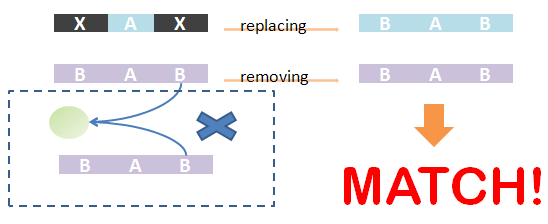 Removing unmatched chains and associated trees of the species. It should have same structure with template species after replacing ST part with matchings if it has the same structural pattern with the template one.
Removing unmatched chains and associated trees of the species. It should have same structure with template species after replacing ST part with matchings if it has the same structural pattern with the template one.
The basic idea is simple. We first consider the structural pattern matching between one chain and a chain template. There are two kind of parts on a chain template: substituent-type part (ST) and non-substituent-type part (NST). The whole template chain could be separated into blocks of ST parts and NST parts. Then we could break down the task to the matching of each small block. For each NST type block, we find all its matching regions on the chain to be matched and there may be more than one regions. Each of the combinations of all NST blocks forms a NST frame and restricts the matching regions of ST blocks. Therefore, the task is reduced to find matchings of each ST part block within a restricted region on the chain to be matched. At the present version of MoDeL, there are 6 ST part: ANY, ANYUB, NZ, NZUB, ONE and ONEUB. A general algorithm for all 6 type part is designed and we will illustrate this via an example. Consider a ST block with all ANY type parts (we use X to indicate ANY type part in the figures). We find matchings of the first part of the block. There may be many possible matchings because part of type ANY implies arbitrary matching of parts on one chain in MoDeL definitions. For each matching found, we deal with the next, and do the same thing recursively until the end of the template or matching fails. An easy example is shown in the figure to illustrate this simple idea.After all possible matchings for each chain in the template have been found, we could choose any one from each to form a combination. In each combination, matchings of different chains should not be the same. For example, there are two chains in the template, T1 and T2, and three chains, C1, C2 and C3 in the species. If T1 has one matching to C1 and one to C2 and T2 has one to C2 and one to C3, the possible matchings are (T1,T2)->(C1, C2), (T1,T2)->(C1,C3) and (T1,T2)->(T2,T3). (T1,T2)->(C2,C2) is not allowed. The general matching relations could be written as (n1,n2,n3...)->(m1,m2,m3...), where (n1,n2,n3...) represents chains in the template and (m1,m2,m3...) represents the chains matched in the species for (n1,n2,n3...) respectively. However, it is only part of the matching relation because details, such as the matching positions of parts in the template, are ignored. The next question is: how to validate each matching?
As defined in Chain-Node model, details of distribution of binding are stored in the forest. Matching of chains could not solely determine the overall match -- whether the template and the species have the same structural pattern. However, it is not easy to compare tree structure directly. We designed an algorithm to do this based on the principle: If the template and species have the same structural pattern, it must be possible to isolate the template from the species after replacing the ST part with its matchings. We first replace the ST part of the template with its matchings in the species. And then, we remove the chains which are not matched and the associated trees -- containing leaf nodes whose corresponding parts are on those chains. If the species after removing has the same Chain-Node structure with the template after replacing, the template and the species have the same structural pattern and in brief, the template could be matched to the species. We also illustrate this idea in figures.
However, there is one problem of this algorithm. Consider a LacI dimer template, whose Chain-Node model has two chains with one part on each and one tree with 2 leaf nodes. And there is also a LacI dimer species, it has the same structural pattern with the template. The matching should be (T1,T2)->(C1,C2) and (T1,T2)->(C2,C1) according to the algorithm described above. However, there is indeed one matching since the two matchings are the same. The underlying reason is that the two chains of the template are equivalent: it makes no difference to the tree structure (Huffman code of each node) if they are exchanged. Hence, several equivalent chains should be matched to a certain set of chains in the species only once.
The algorithm of species matching is of the most importance since it provides technical support for structure-based description of species and reactions. It costs most of the running time. We will try to increase the efficiency of the codes of this part.
Generation of products based on reaction templates
Within MoDeL definitions, product templates are composed of both ST parts and NST parts. The ST parts are all transfered from reactants or modifiers. A transfer table is defined in each reaction template to label where does each ST part comes from and goes to. It seems to be very easy to generate Chain-Node structure of products from reactants and mod modifiers based on the transfer table. However, it doesn't.Consider first a template reaction with one reactant, X-A-B-Y, where A and B are both NST parts and X and Y are both ST parts with type ANY. If it could be digested by a certain enzyme, the products should be X-A and B-Y. In the real biological system, if there is a species with structure C-A-B-C, where the first part C and the last C binds to form a tree node. If we try to digest the A-B bond, the species would not be split into two pieces as expected because the two products are connected by the tree node. To generate the structure of products correctly, we design a three-step mix-trim-split algorithm to solve this problem.
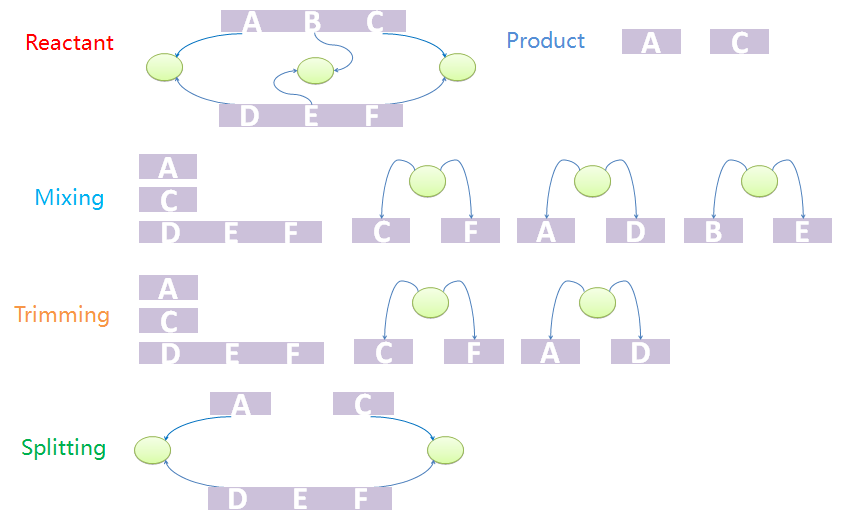 Mixing, trimming and splitting: three-step algorithm to generate products.
Mixing, trimming and splitting: three-step algorithm to generate products.The first step is to mix reactants and products. The mixture includes all chains of reactants except for those which are matched to the templates as well as all trees. It also includes all chains and trees of products. And now we have a mixed species with chains and trees from different sources. It is worth noting that chains and trees of modifiers will not be added into the mixture. The next step is to trim the trees. Trees with leaf nodes that are not defined in chains will be removed from the forest. The last step is to split the products out from the mixture. This is because the mixture may not be one standalone complex. For example, it may have two unbound and uncorrelated chains. In other words, some chains that are not connected with others via tree nodes should not be incorporated into other species. The final standalone split species are real products in the system.
We provide an example to illustrate the product-generating procedure. The reactant has two chains, A-B-C and D-E-F. A, B, C bind with D, E, F, respectively. The products are A and C. First we mix chains and trees. Chain A-B-C will not appear in the mixture. Since node B does not appear in mixed chains, tree with nodes B and E will be removed in the trim process. Because part C and F, A and D, are both bound to nodes, the final product should contain A and B both after split processing.
In the example above, there is only one product finally though two product templates are defined in database. What compartment does the product locate in if the two product templates locate in different compartments? A reasonable solution is to halve the quantities of the product and add it into both compartments. Another problem is that the corresponding reverse reaction would never be found because it fails to match two templates to only one species. The source of the two problems is essentially the same: how to describe intra-molecular reactions in framework of MoDeL language definitions. At present, it is not supported. However, supporting intra-molecular reactions is one of our future plans to refine our MoDeL language.
- Resources
Resources
On this page we provide the information of downloading the executable files and source files of our project, and the documentation to introduce the source code build steps and system prerequisites.
Downloads
Executable files can be download at SourcForge
Project source code is available at gitHubSource Code OverView
We use C++ as our programming language, cmake to manage our project, git to reversion control, yacc and lex to build our model file input assistant tool, Qt4 to build the GUI, Oracle dbxml as our Database manager system. Other wise, several 3-rd party open source software or tools are used in our iGame source code. Libsbml is used to deal with sbml file operations. Qwt and Qwtplot3D are used for simulation data visual system. At the early stage of our project SBML Ode Solver and Sundials were our main ode solver to produce the simulation data from our sbml model file, because the latest SBML Ode Solver is 1.6.0, developed in 2005, which is out of date and fail to support the newest sbml Level 2 Version 4 standard. We succeed in updating the SBML Ode Solver 1.6.0 to support SBML L2V4, but some unfixed bugs was found, so we turn to a better ode solver copasi, which moreover can flexibly display the ode solver output data and modify parameter to adjust the simulation output data curve. Obviously, those open source library and tools convenient to our design and implementation and help us to focus on our core working.
Build instruction
Prerequisites
To sum up, iGame should be complied and run in computers with following tools and libraries:
- C++ development environment. In Linux should be GNU C++ Tool Chain And in windows MingW Tool Chain.
- Qt4 develop and run-time environment. Qt4.7 is requirement. Editor or IDE like QtCreator or Vim is also needed.
- CMake, to configure the project and generate the Makefile. CMake 2.8 or later version is a requirement
- dbxml, The Berkeley xml native database, is available at http://sourceforge.net/projects/dbxml-core/
- copasi, copasi 4.6 is a requirement. the source code and executable binary file can be downloaded at http://www.copasi.org/tiki-index.php?page=download, and one can follow those instruction to build it from source code. What should be noticed is that copasi also has prerequisites, including LAPACK/CLAPACK for Maths, Qwt and Qwtplot3D for plotting, expat to parser xml, raptor The latest version of this RDF parsing library, and libsbml for deal with SBML operations.
- libsbml, Version 3.4.1 is required. Not only it is the dependence of copasi, but also the dependence of igame core code.
- Bison & Flex
- git, to download our source code by command our source code can be downloaded in github, git is helpful to download.
- git clone git://github.com/jkdirac/igame.git
Build
We use cmake to manage the build progress, cmake can generate cross-platform Makefile, we intend to make our project could be built in different operating system. Until now, only Linux operating system is tested. Windows and Mac should be supported soon. Download all source code, run cmake-gui to configure the project. Pay attention to the configure information.There should be "missing ***" messages when Cmake fail to find the dependent libraries. To fix those messages, one need to assign the path of related libraries by Cmake GUI. When Makefiles are successfully generated, execute the Make command to build.
- Overview
-
One-Minute Introduction
You are about to begin an exciting journey of the main ideas in MoDeL. For each picture, start from the green flag, follow the arrows, and finally you will reach the destination! Read on from the first step or click one of the three steps!
Chain-Node Model -> Template Modeling -> Automatic Modeling Database Language
Chain-Node Model
Template Modeling
Automatic Modeling Database Language
- Features
-
Features
Chain-Node Model
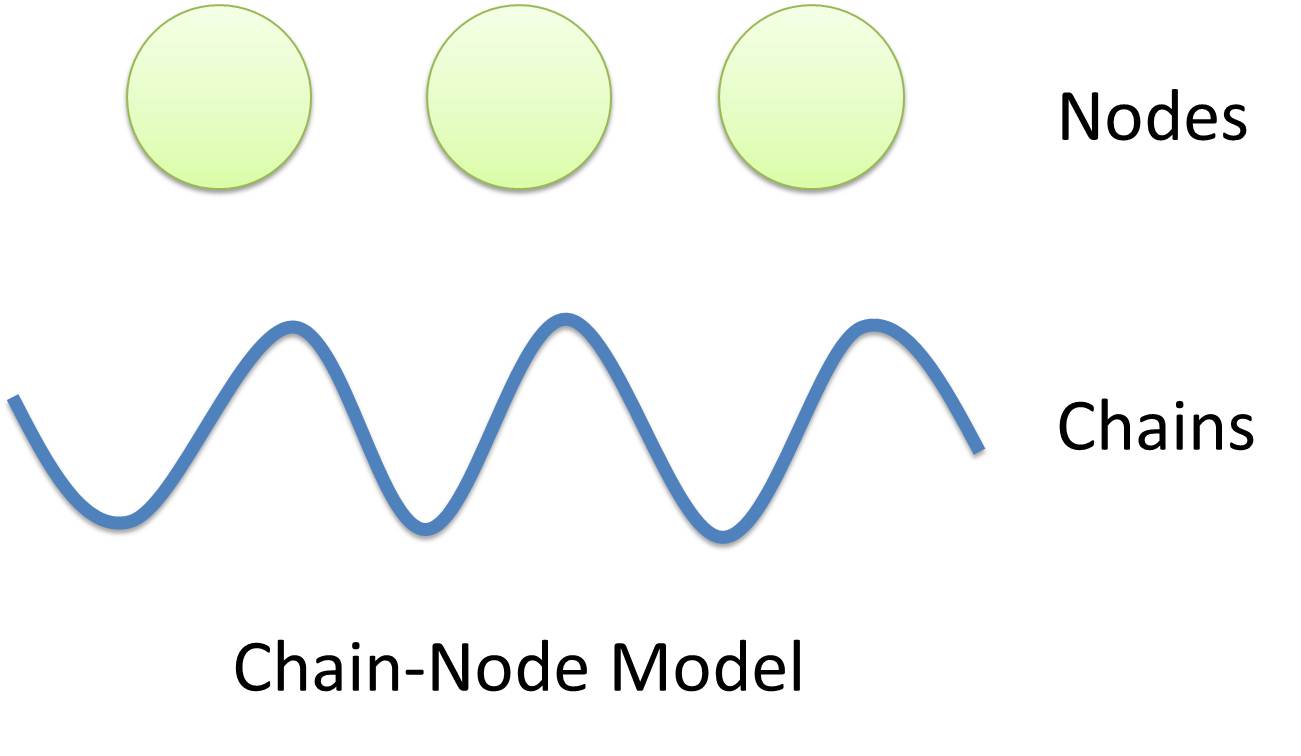 Figure 1: Logo of Chain-Node Model
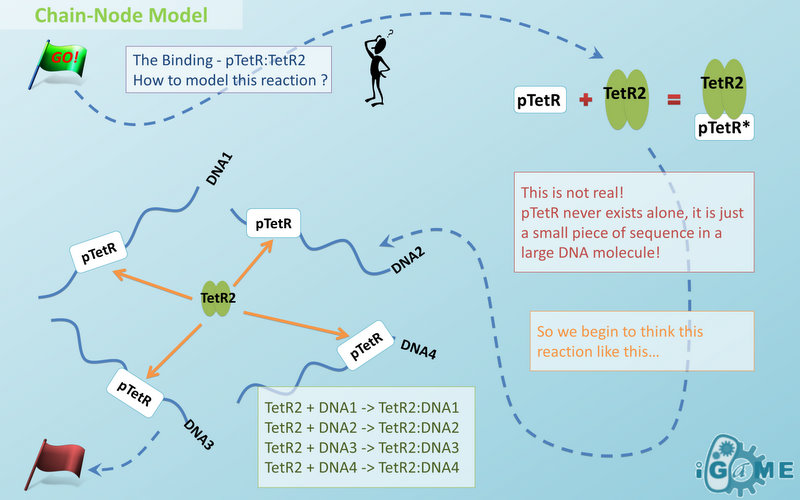
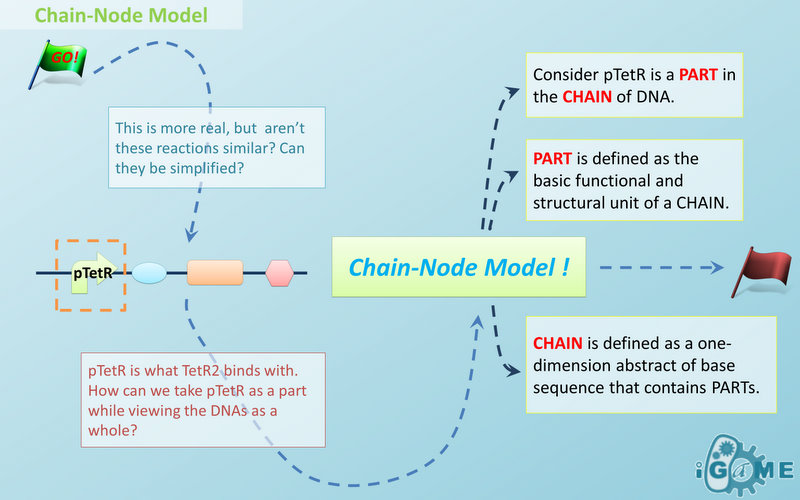
Figure 1: Logo of Chain-Node ModelIn most manual modeling, complex is treated as a whole entity in reactions. Plenty of synthetic biology automatic modeling software treats complex the same way. However, there is a big difference between manual and automatic modeling, that is: Model constructors always know which detail of complex needs to be ignored or preserved, but software does not. So in most software, detailed structure has to be specified by modelers, otherwise will be ignored. In most cases, this requires plenty of manual operations and thus making the software not so automatic. Knowing this, MoDeL chooses to preserve detailed structure in complex and present Chain-Node Model, which view complex as a construction of its parts throughout the modeling process. With chain and nodes, describing large biological complex with a great number of basic units and binding sites becomes possible. It also offers great flexibility in modeling. Detailed concepts are explained below.
Abstract Concept of Chain
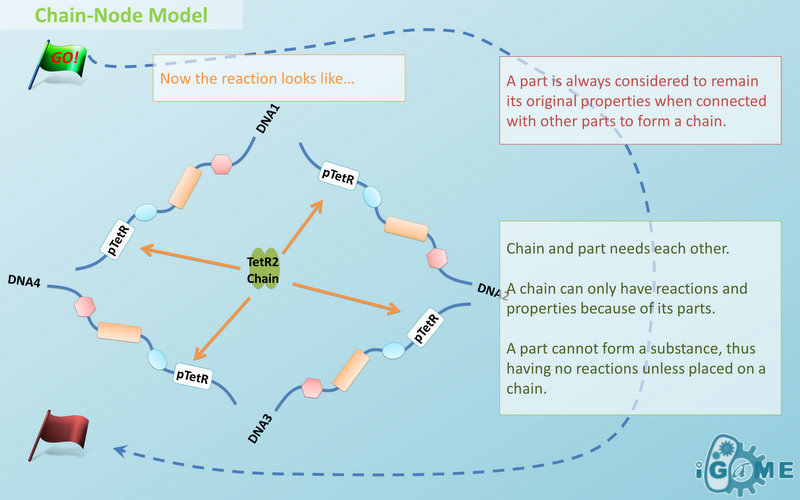
When it comes to biobrick assembly, DNA of plasmid is often considered as an abstract straight line where biobricks can be placed on and connected together. It actually acts as the basic frame of biobrick assembly. Inspired by this, we present the abstract concept of chain, which also acts as the basic frame of part assembly. Chain is an abstract straight line where different parts can be placed on and connected together. Part is the basic unit of chain, and chain can have as many parts as possible. To go beyond DNA, and to unify the data structure of Species, we extend the concept of chain in MoDeL to include all the parts we can describe, including RNA, protein, and even compartment, compound and substituent, etc. Parts of DNA, RNA and protein, are all originated from DNA, and they are considered as “Sequence”. Sequence can be connected to other parts of sequence on a chain. On the contrary, parts of compartment and compound are considered as “Non-sequence”. Parts of non-sequence cannot be connected to any other part on a chain, which means, they can only exist on a One-Part-Chain. Substituent is used as a substitution of any other part, so it can be either sequence or non-sequence. It is important here to mention that the Sequence/Non-sequence division is only a rule guide the modeling. It is not a constraint to Chain-Node model. Actually, we choose to maximally preserve the flexibility and leave all the freedom of constructing a species to users. Users are allowed to construct ANY species they can using Chain-Node Model. For example, one can create a strange species with compartment, compound, and DNA parts on the same chain, which makes little sense in biology. So, it is really up to the users to decide how to use Chain-Node Model in modeling.
Abstract Concept of Node
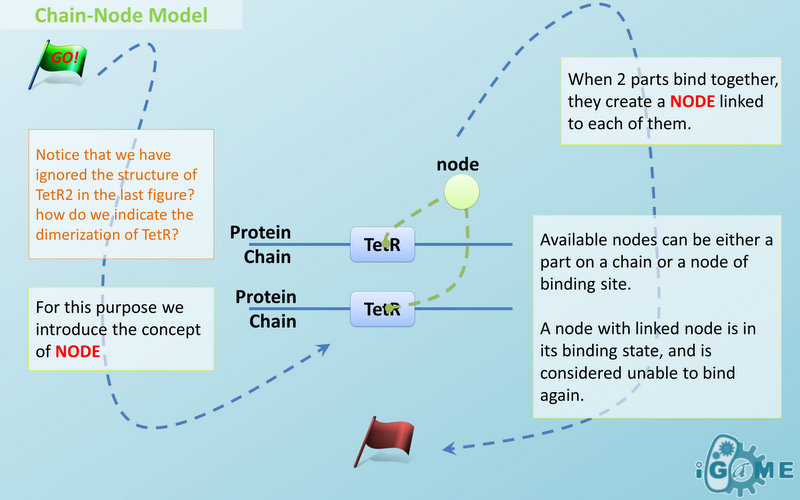
Node is used to characterize binding structure in biological complex. Node is an abstract concept of binding site or site that can be bond. Links always exist between a binding site and the sites it binds together, indicating the relationship between them. We use trees to represent the binding structure in complex. The parent node represents the binding site, and the child node represents the bond sites. For example the binding structure of TetR dimer is considered as a TetR2 node with two TetR nodes as its children. There is one exception though, the ROOT node. ROOT node is used as the top node in a tree. It can only have one child and no parent. The function of ROOT is to indicate that its child is the last binding site in this tree, and it binds to nothing any more. In the reactions of promoter repression and activation, the promoter is repressed or activated after binding with repressor or inducer. So its properties are changed, and it is not the original promoter any more. This is an example where binding changes the properties of node. Since we do not store all the possible property alterations within one part, the solution is to replace the old part with a new part that stores the correct properties. Available nodes can be both a part on a chain or a node of binding site, which leaves a lot of possibilities in constructing complicated binding structure. Again, there are no restrictions, but parts of compartments are not suggested to appear in a binding structure.
The Basic Assumption
The basic assumption of Chain-Node Model is that a part ALWAYS carries its properties wherever it is placed in an instance of Chain-Node Model. This means no matter how complicated the complex is, you can always keep track of the location and properties of every single part. By letting complex inherits its parts’ properties, the assumption saves modelers from rewriting the structure and function of each new complex. However, we should address that the assumption is not suitable for all circumstances. For example, under this assumption, a fusion protein will carry the properties of the fused proteins, which is not true in many cases. A fusion protein may not remain its old properties and even have new properties that none of the original parts have. In this case, our suggestion is to define the fusion protein as a new part. So it is suggested that users choose different strategies of describing the same thing in Chain-Node Model base on different situations, instead of modeling everything in the same way.
Examples
With great power there must come great responsibility. In order to use the powerful Chain-Node Model correctly, please have a look at a few examples we presents. Example of a species: E.coli cell In MoDeL, the cell of E.coli is described as a species with only one part of E.coli(see Figure 1). Since the concept of part has been extended greatly, living cells can be described in the same way other species. Compartment belongs to “Non-sequence”, so it is suggested that E.coli only exists as a one-part-species.
 Figure 1: The Chain-Node Model of a E.coli cell
Figure 1: The Chain-Node Model of a E.coli cellExample of a species: pLacI*:LacI4 and its tree structure of binding. Here is another example of template species with more complicated structure.(see Figure 2) In biology, LacI tetramer (or LacI4) can bind with promoter pLacI as a repressor. When bond with LacI4, pLacI is repressed and its transcription speed will decrease significantly. In order to express this alteration, as mentioned above, we use another part of pLacI* with decreased transcription speed instead of pLacI in the species.
The structure of binding sites is shown in figure (see Figure 3). The graph indicates that LacI monomers first bind together to form LacI dimer, then LacI dimers forms LacI tetramer, and finally LacI4 binds with pLacI* to form pLacI*:LacI4. 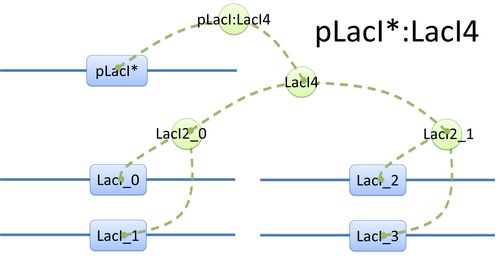 Figure 2: Species of pLacI*:LacI4
Figure 2: Species of pLacI*:LacI4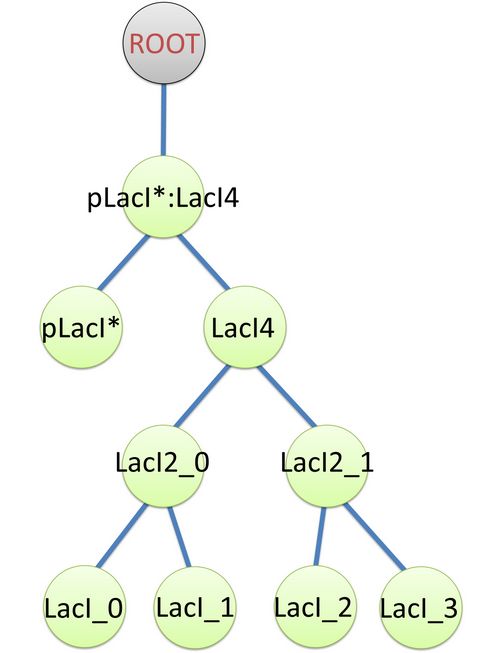 Figure 3: Tree structure of binding in pLacI*:LacI4
Figure 3: Tree structure of binding in pLacI*:LacI4Modeling with Templates
Manual modeling of biological system is widely used in synthetic biology. However, it requires an overall understanding of the biological network and large amount of data, making it difficult even for professionals. Actually, the data provided manually are redundant because different reactions may occur with a same mechanism. Based on this idea, we introduce the concept of Template, which is the minimal data set for describing biological reaction networks, as a feasible way to implement automatic modeling. The term “automatic” does not mean modeling without providing any information. Instead, we aim at providing minimal amount of data. Similar to C++ programming language, templates allow generic description of species of a certain structural pattern or reactions of a certain reaction mechanism. Substituent and Template
There are two kinds of templates: Template Species and Template Reactions. They are designed to represent a family of species with similar structure and a family of reactions with similar mechanism respectively. In order to realize this idea, we add the container of Substituent into MoDeL. There are currently six objects in this container, ANY, ANYUB, ONE, ONEUB, NZ and NZUB. Though defined differently, they actually serve the same purpose: representing other parts. Let’s take ANY as an example (see Figure 4). ANY, represents parts on ONE chain and the number of represented parts can range from zero to infinity. For example, a DNA species with structure ANY - pTetR - ANY represents any DNA with part pTetR. 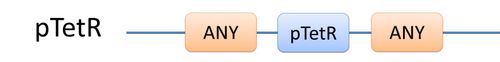 Figure 4: Template DNA species
Figure 4: Template DNA speciesA reaction with template species as its reactants, modifiers or products is a Template Reaction. A template reaction describes the core structure of reactant that causes the reaction to happen and provides a specification for generating reactions with the same mechanism. This could be understood more clearly by dividing parts in a species into functional groups -- a reaction template describes the interaction mechanism of these functional groups. For example, pTetR promoter is deactivated in presence of TetR dimer. The template species are pTetR DNA template and TetR dimer protein template (see Figure 5) and the functional groups are pTetR promoter and TetR dimer. All the parts of ANY should also be counted as a functional group, though they don’t have functions in this reaction. Any pair of species which partially contain pTetR DNA and TetR protein dimer respectively would bind according to the description of this reaction template. Modeling with templates allows users to define species and reactions only once for one certain family without rewriting them again in database. 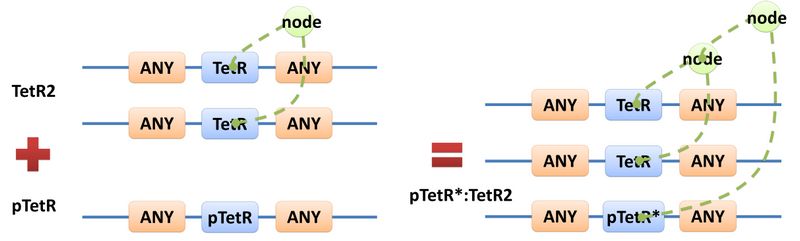 Figure 5: Template reaction
Figure 5: Template reactionWe are still looking for more substituents. We are planning to have more detailed substituents that can replace certain number of parts on certain number of chains. Even the arrangement of parts and chains can be specified. We believe it will make our template modeling more powerful.
A Standard Language
MoDeL aims at providing a language and syntax standard for automatic modeling databases used in synthetic biology models. We will discuss a few relevant topics in the following sections. Standard Biological Part Automatic Modeling Database Language
With plenty of information for automatic modeling needs to be stored, we design our database, and develop a standard language of describing the database. We call the language: MoDeL. MoDeL (Standard Biological Part Automatic Modeling Database Language) is a database representation format for synthetic biology automatic modeling. This project of MoDeL aims at providing a language and syntax standard for automatic modeling databases used in synthetic biology models on a number of topics, including cell signaling pathways, metabolic pathways, biochemical reactions, gene regulations, and so on. MoDeL is developed as a machine-readable format in XML fashion, programming languages with XML library API (such as libxml2 for C++) are strongly recommended for reading MoDeL database. USTC_Software has been devoted to the standardization of MoDeL. Documents can be found here. Example of database written in MoDeL can be downloaded on our GitHub page. MoDeL is an attempt to define a universal database markup language for synthetic biology automatic modeling. It supports descriptions of a wide variety of different parts, including biobrick, compound, compartment, substituent, and so on. It can also describe the species constructed by these parts, and reactions happen with the species, such as binding of species, dilution of species, degradation of proteins, amplification of cells, multi-compartment reactions, and so on. Many traditional methods fail to consider structure of species when trying to markup reactions with only reactants, modifiers, products and kinetic laws. The best part of MoDeL is that it can preserve detailed structure in species and reactions using Chain-Node Model. Based on the model, MoDeL is able to markup almost all kinds of reactions in a detailed and clear way. Overview of MoDeL
Figure 5 above is an example of a simple network of biochemical reactions(pTetR + TetR2 = pTetR*:TetR2) that can be automatically constructed using MoDeL: MoDeL supports automatic construction of such network with only the input of its initial conditions and environmental parameters. In order to do this, obviously the data of parts, species, reactions, compartment, units and so on is needed. Besides, relationships between these components are also needed, so we can link to a reaction when we find a species. Data of relationship is stored as references and exists in most MoDeL components. To define all the needed components in a systematic way, MoDeL organizes them in five component containers at the top level: :: System: This container contains definitions of units, functions, rules and global parameters. These are all basic elements for other components of MoDeL, and many are mathematical concepts without direct synthetic biology meanings. The name of the container, System, separates this container from others which contains components of biological significance, and indicates this container includes the basic parameters of the whole system. :: Compartment: This container contains definitions of compartments. Compartment here is considered as a holder of substance with certain volume, so it only stores related data such as units and volume. It also stores a list of compartments that are permitted to be placed inside. For compartments like E.coli that can multiply, a corresponding compartment with the same id will be created in the sub-container of compartment in Part. :: Part: This container contains definitions of parts. Part includes the basic units that can be inserted in an abstract chain. The container of Part has five sub-containers which are: compartment, biobrick, compound, plasmidBackbone, and substituent. Biobrick contains biobricks from PartsRegistry together with other similar components created by users. Compartment contains cells and other microscopic organisms that can multiply. compound contains small molecules like IPTG and other compounds. PlasmidBackbone contains plasmid backbones come from PartsRegistry or defined by users. Substituent contains substitutes for template modeling. :: Species: This container contains definitions of various species defined by Chain-Node Model. Each component stores the cnModel structure of chains and trees. Related reactions are also stored. It is suggested that the constructor of a database stores template species instead of real species in this container. Only in this way can he make use of the power feature of template modeling of MoDeL. :: Reactions: This container contains definitions of various reactions. Each component has its own compartments, reactants, modifiers, products, substituent transfers, and kinetic law. This forms a detailed description of reactions. Again, constructor of a database is suggested to store template reactions instead of real reactions. The organizational structure of components in MoDeL is shown in the following figure. 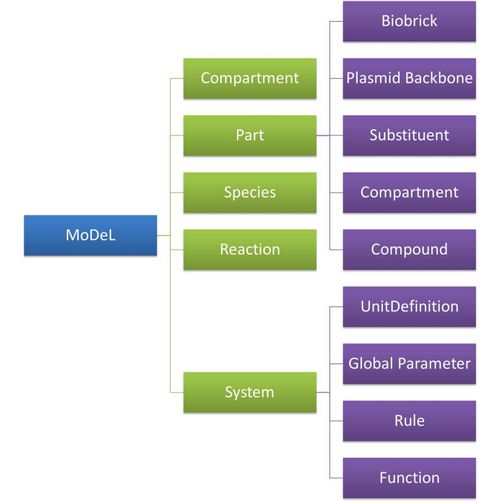 Figure 6: Structrue of MoDeL components
Figure 6: Structrue of MoDeL componentsAs mentioned above, reference relationships are stored in most components. The following figure shows the relationship between different components, the pointer pointing to a component indicates referencing to that component. 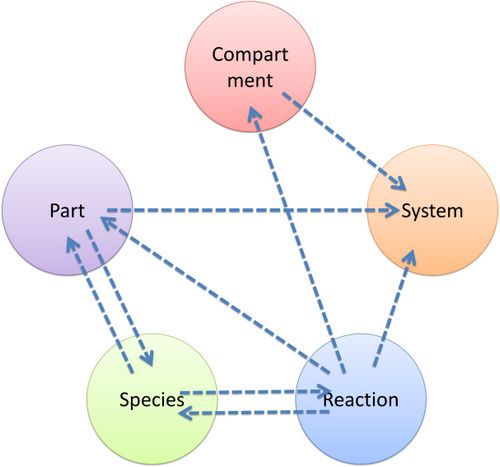 Figure 7: Reference relationship of MoDeL components
Figure 7: Reference relationship of MoDeL componentsIt is worth noting that MoDeL does not attempt to include all information about each component. In PartsRegistry, a long list of data in XML format is provided including part author, status, sequence, and even group access information. Such data are redundant for our database since they are not necessary elements needed to construct reaction network. Only the useful data is retained. Relationship with SBML
SBML(The Systems Biology Markup Language) is a machine-readable language in XML fashion. It's oriented towards describing systems where biological entities are involved in, and modified by, processes that occur over time. As a widely used model exchange standard, SBML is chosen to be the output of our auto-modeling module. Generally speaking, MoDeL needs not to be conformed to any constraints set by SBML. However, since SBML is a widely used standard, it is better for MoDeL to share common concepts and components with SBML as many as possible. For example, MoDeL still uses rules, function definitions and unit definitions in SBML. By doing so, it will be easier for software tools to change formats between MoDeL and SBML. - Language Specifications
-
Standardization of MoDeL

MoDeL aims at providing a language and syntax standard for automatic modeling databases used in synthetic biology models. Knowing the importance, USTC_Software puts a lot of effort on the standardization of MoDeL. We have requested BBF RFC55 for MoDeL, and constructed a beta version of XML schema to describe the XML structure of MoDeL.
BBF RFC55
BBF RFC55: Standard Biological Part Automatic Modeling Database Language (MoDeL) mainly describes the detailed language specifications of MoDeL with concrete examples, which provides enough information for serious database constructors to build up a database on his own. As a highlight of MoDeL, the concept and the syntax of Chain-Node Model are described in detail. The content of document is currently arranged as follows:
 Figure 1: Draft document of RFC55 - Table of Contents
Figure 1: Draft document of RFC55 - Table of ContentsThe document of RFC55 has already been submitted. You can find it on the webpage of The BioBricks Foundation:RFC. Following are some more previews at the draft document.
 Figure 2: Preview at draft document of RFC55
Figure 2: Preview at draft document of RFC55 Figure 3: Preview at draft document of RFC55
Figure 3: Preview at draft document of RFC55XML Schema
XML Schemas express shared vocabularies and allow machines to carry out rules made by people. They provide a means for defining the structure, content and semantics of XML documents. Being written in XML fashion, a Schema is perfect for MoDeL to allow software to perform check on syntax. Following is the preview of the beta version of XML Schema for MoDeL. The Schema can be downloaded at iGaME project page on SourceForge.
 Figure 4: Preview at XML Schema of MoDeL
Figure 4: Preview at XML Schema of MoDeL Figure 5: Preview at XML Schema of MoDeL
Figure 5: Preview at XML Schema of MoDeL
Monthly Notebook
March April May June July August September October Daily Notebook
March M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 April M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 May M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 June M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 July M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 August M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 September M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 October M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 As a tutor game aimed at improving the understanding and facilitating IGEM and synthetic biology, iGAME itself is exactly a great way of human practice. With iGAME, people do not feel any distance to synthetic biology any more. Actually, it is time for everyone to join this trend to be as cool as Sheldon’s new biologist girlfriend. However, we do not stop here. We paid great effort to the safety issue and we have been dedicated to improving the knowledge of synthesis biology from classes and tried to share the part we contribute to the worldwide. We innovatively plot a new way of spreading the spirits of IGEM by starting our own community and we will never stop sharing and communicating.
--Luojun Wang, author of USTC_SOFTWARE human practice
Human Practice,"the C project"
Although we do software, we also keep eyes on promotion of synthetic biology and sharing our own program. Therefore, we started a long term human practice, “the C project”, since this summer.“the C project”, as we call, consists of three parts: Curriculum, Communication and Community.
Curriculum
As a IGEM team, we try to let people as many as possible to know what synthetic biology is. Therefore, advisors of our team, Haiyan Liu and Jiong Hong, have established a set of synthesis biology curriculum this summer. We have done many works related to human practice on the curriculum.- Jiong Hong (in the right picture), advisor of our team, gave lectures introducing safety and ethnics issues of synthetic biology.
- Luojun Wang (in the head picture), members of our team, gave lectures promoting iGAME.
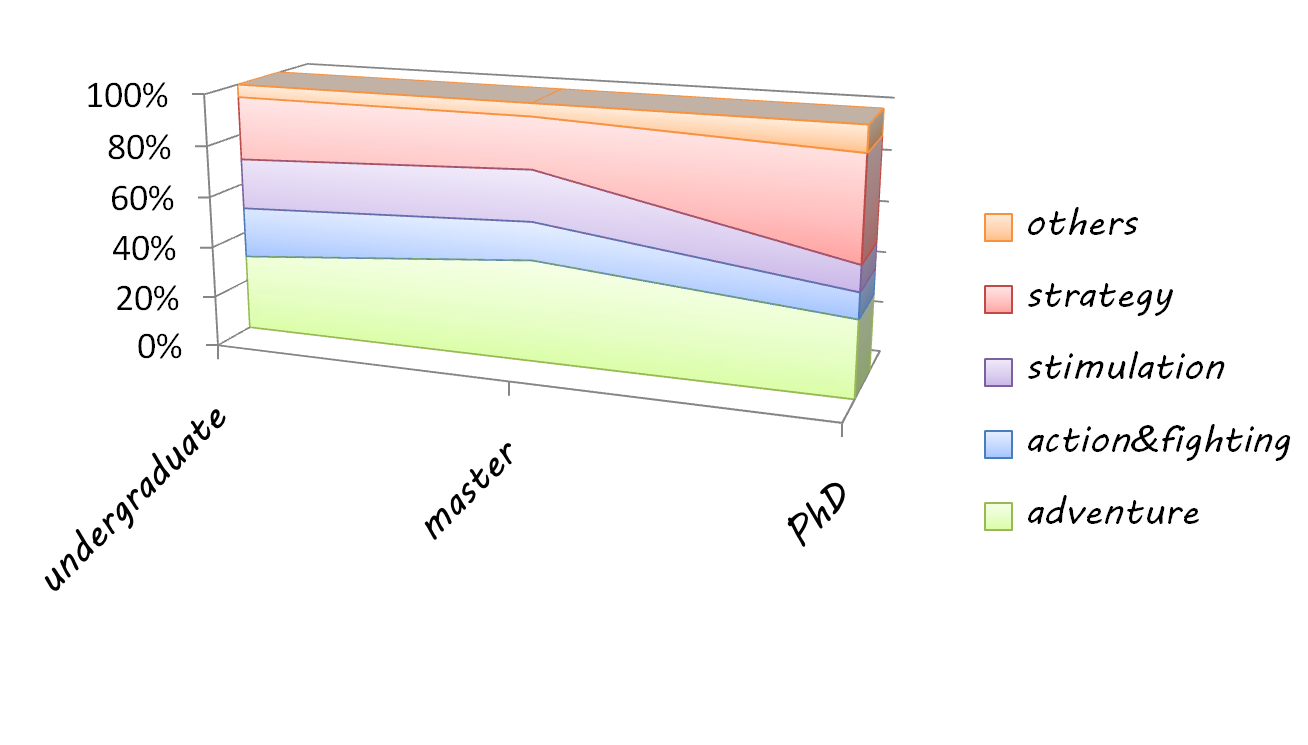
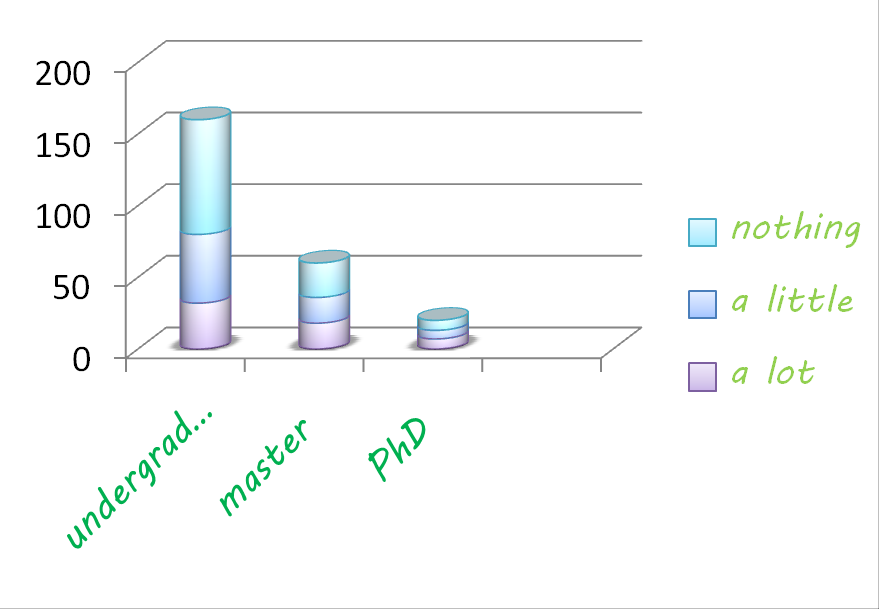
- A survey* on how students know about synthetic biology and their interests for tutor game was made.(*the survey statistics comes from a sample of 240 students. Numbers of undergraduates, masters and PhDs are 160, 60 and 20)

Analysis of the results of the survey The first chart shows students' favorite game types for iGAME, and the second chart shows their knowledge of IGEM project.
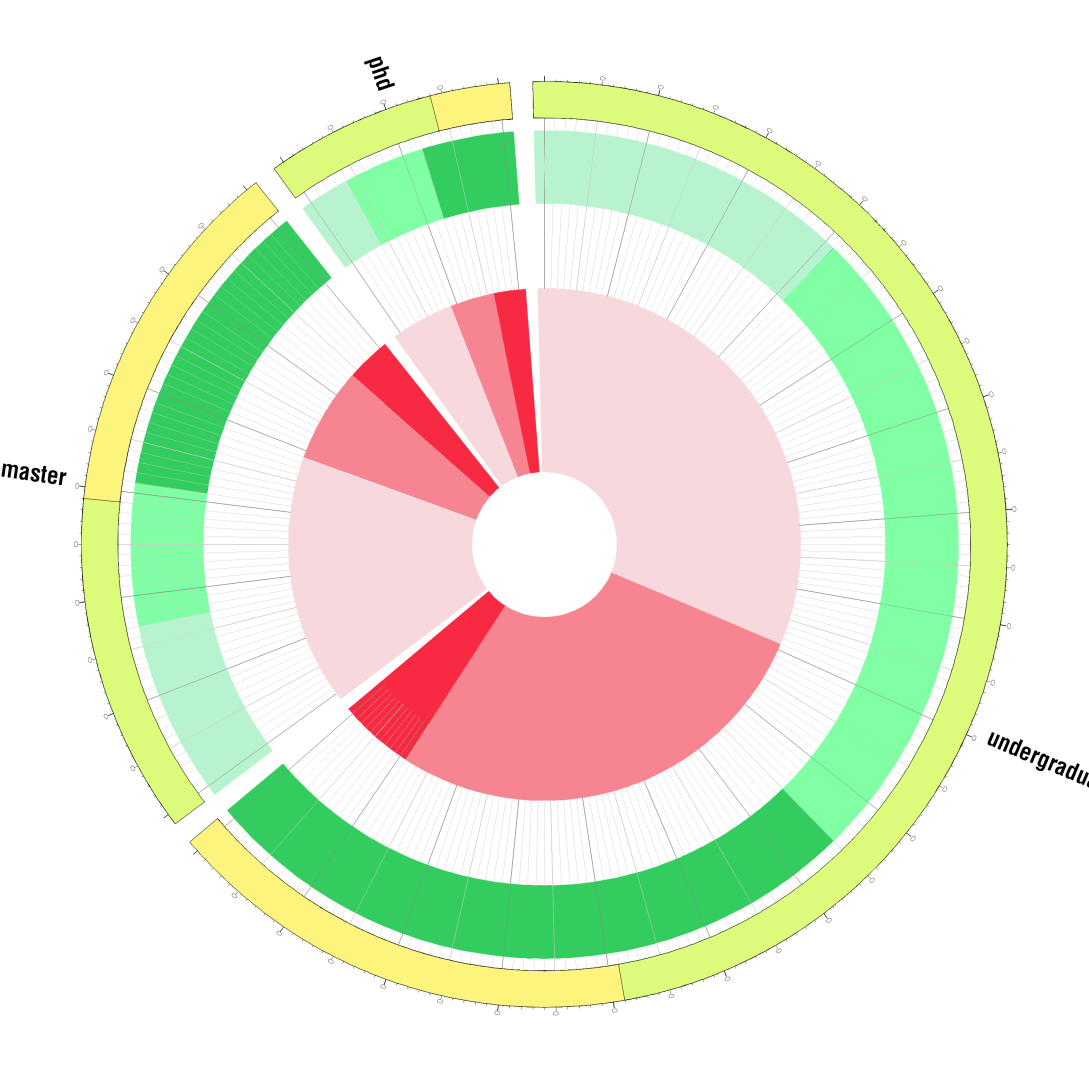
The Circos Map
We tried many methods to plot our survey more straightforwardly and clearly before we finally chose Circos mkweb.bcgsc.ca/circos. Circos is biological software used to plot genome elements and relationship. Here, we innovatively used Circos to demonstrate our survey.(as in the right picture) Circos is somehow like pie chart yet it can present multi information as all the statistics are proportional and clear on the graph.In the graph, there are three circles. The inner circle stands for their knowledge of synthetic biology, and middle circle stands for their interests of iGame. Deeper the color is, the more they know of synthetic biology or the more they are interested in iGame. For example, light pink means they know little about synthetic biology and deep green means they are very interested of the coming tutor games. The outer circle, however, stands for their major;light green for biology major and light orange for other majors. As the graph shows, synthetic biology is still new to them as the majority have little idea of it, for which is what we will continue our project in the future. However, we are delighted to see that their interests of tutor game are fairly strong.
Referring to the possible game types, adventure and strategy games are students’ favorite, while some of them prefer stimulation and action games. In our program, we tried our best to listen to their voice and develop a game which is both educational and appealing.
Communication
We’d like to share what we have achieved with the world, so we did and will do a great deal to communicate with others and learn. For example, we collaborate closely with USTC wet team to learn what biology majors think of iGame and improve. Several seminars have been hold to make sure our program can really help the promotion of both synthesis biology and our tutor game. In addition, further transnational communication is not absent, like we had a deep talk with Dr. Joy Yueyue Zhang*(*BIOS Centre, London School of Economics and Politics Science) on how our program can change the development of both IGEM and make biology a better subject.Yet we do not stop here. We have sent emails to other teams for further communications and asked for their opions on how we can improve iGame. What is more, all our codes are shared on github[1](in the right picture). With fixes and new features putting forward every day,any one in any corner of the world can learn and keep update of our work, not just from the surface, but from codes and essense of our work. This is really a great way of communication, for we do not have to say too much to put our achieves across to those who really want to know our work, which really works. Great contributions have been given through github and we get a lot of advices and opinions as well.
Community
To better promote synthesis and the spirit of IGEM, we decided to establish a community together with IGEM USTC team. For the community, we have set regular classes and lectures for both synthesis biology and IGEM. We also open our laboratory to our community members and teach them basic skills of biology experiment(as in the right picture), like PCR. Of course, this is the main field we will update our progress on in the future.
More information of our community and other human practice of USTC IGEM team, please click2010 USTC IGEM humanpractice!
- Overview
-
Team
 Team
TeamThis year 10 students start USTC_Software igem team. Chen Liao, Zheng Wang, Kun Jiang, XiaoMo Yao, Jue Wang, Luojun Wang, Yubo Qi, Zhaoyi Li, Zhengda He, Shangyu Luo. There are only 8 of us in the picture, when the picture was taken the only female team member, Jue Wang was in Beijing for her TOEFL exam. And Shangyu Luo had been to HongKong for studying in The University of Hong Kong as exchange student. What a pity! But they will join us at igem 2010 Jamboree!
USTC has two IGEM teams this year, One wet lab team and our software team, all students majoring in biology was attracted to the wet lab team. Our team consists of students of other departments, such as Mathematics, Physics, Computer Science and so on. Thanks to our Instructors, Hao Jiang, Zhaofeng Luo. With their professional instruction, we are guided to decide the project plan, design and implementation.
About iGEM@USTC
The iGEM Team of USTC are the combination of the exact tradition of USTC and the exact vitality of the young. Rigorous daily schoolwork has provided the team members with a solid foundation of basic knowledge, and an innovative spirit enables us to stand on the frontier of the modern science.
In 2007, Jian Zhan, Bo Ding, Rui Ma, Xiaoyu Ma, Yun Zhao, Ziqing Liu and Xiaofeng Su, guided by Prof. Haiyan Liu, Prof. Jiarui Wu and Prof. Zhonghuai Hou, won the "Best Foundational Technology Prize" and a Gold Medal, with the project "Extensible Logic Circuit in Bacteria".
In 2008, Xunyao Wu, He Shen, Genxin Liu, Jia Liu, Binjie Xu, Yanting Xue, Yu Chen, Zhen Xu, Bo Ding and Jian Zhan, guided by Prof. Haiyan Liu, won a Silver Medal, with the project "Artificial Multi-cellular Self-organization System".
In 2009, Hao Jiang, Li Xing, Hao Wu, Zongxiao He, Danqian Liu, Chao Li, Jiayi Dou, Hanyu Lu, Hao Zhou and Bing Wu, guided by Zhaofeng Luo, Jian Zhan, Jiong Hong and Xiaoxiao Ma, won a Gold Medal, with the project "E. coli Automatic Directed Evolution Machine (E.ADEM)".
In 2009 we also formed a software team for the first time. Wei Pan, Yuwei Cui, Jiahao Li, Yu He and Xiaomo Yao, guided by Bo Ding, also won a Gold Medal, with the Project "Automatic Biological Circuit Design (ABCD)".
Since last year we have accumulated some experience from the project of Automatic Biological Circuit Design (ABCD), we expect to develop a synthetic biology game this time. The game aims at teaching non-biologists how to design biological systems so that players can compete to improve their designs in order to help biologists create the best engineered machines!
For more information, go to USTC iGEM Forum.
About USTC
- Message from the President
A university is the important assurance of maintaining belief, developing science and culture. It is a significant symbol of the level of civilization achieved by a nation.
- USTC Today
The University of Science and Technology of China is a new type of university established by the Chinese Academy of Sciences (CAS) after the founding of People's Republic of China. The establishment of the University is aimed at fostering high-level personnel of science and technology absolutely necessary for the development of the national economy, national defense construction, and education in science and technology. The University was founded in Beijing in September of 1958. Mr. Guo Moruo is the first president of the University......
- History of USTC
"Pooling the entire strength of the CAS for the development of USTC and combining the institutes of the CAS with the departments at USTC" "Studying hard and excelling in both integrity and specialty"USTC was established in Beijing in September, 1958 under the presidential leadership of Guo Moruo. The establishment was hailed as "a major event in the history of Chinese science and education"......
To get more information, please visit http://en.ustc.edu.cn/ in English and http://www.ustc.edu.cn/ in Chinese.
Team Cooperation
Helps and acknowledges
Gallery
















- Students
-
Students
 Xiaomo Yao
Xiaomo YaoI am soumort! I have nothing to say!!
 Chen liao
Chen liaoMy Profile:
Chen Liao, Room 418, Key Lab of Quantum Information, CAS, East Campus, USTC Hefei, Anhui 230026, China, liaochen@mail.ustc.edu.cn
I’ve just began my first year in the Optics and Optical Engineering program as a master candidate at University of Science and Technology of China. I became interested in iGEM because I found it a great chance to learn synthetic biology. Since iGEM is a student-run and student-manage programe which encourages students both to work collectively in a team and work individually to complete one's own task, it is a great experience to exercise my team working ability as well as creative ideas. In the last months, especially the summer, I worked with students from different disciplines to design and implement our project together and have learnt a lot from them. It is rather a wonderful experience to work with all team members. (^▽^)
As a student of department of physics, my research area is development of first principle theory and implementation of LCAO (Linear Combination of Atomic Orbitals) algorithms. As a member of LiXin He's group, I have successfully implemented atomic force calculation in LCAO method and my next project is to modify and develop algorithms under condition of magnetic B field. Besides scientific research, I do sports very often. Basketball is my favorite and my favorite basketball player is Kevin Garnett (K.G.), his professional attitude on the court as well as his individual character in the life impresses me a lot.
 Zhen Wang
Zhen WangI am Zhen Wang!
Zhen Wang, Room 418, Key Lab of Quantum Information, CAS, East Campus, USTC Hefei, Anhui 230026, China. Email: xqqixp@mail.ustc.edu.cn; wangzhen.ustc@gmail.com;
Hi, I'm Zhen Wang, I mainly work on MoDeL and Database of iGAME. As a member of Lixin He's research group, I focus mainly on calculation of quantum information, quantum correlation and entanglement.
You might have noticed that my address is same as Liao's. Yes, we are from the same Research Group, and the same department, too. It will be boring if I repeat all the information again, so I am going to write some experience of myself participating iGEM2010.
As a student majoring physics, it is hard for me to believe that I can participate in a biology project. Luckily, to my great surprise, these two areas are not so far apart. Modeling and analyzing skills of physics can also be used in biology, and dry project always means a lot of coding task, I can handle that, too. Of course, there is plenty of biology I need to learn, but with the help of our instructor and wet team members, I learn it fast. So I want to tell everyone that there's no limit in iGEM, join in and unleash your power!
 Kun Jiang
Kun JiangEmail: jkdirac@mail.ustc.edu.cn
I am Kun Jiang, the cheif software developer in iGem 2010 USTC_Software team, responsible for designing the global architecture of our software and developing the ODE Solver module, Berkley database interface module and several simple tools for teammates.
It is my second year of studying in computer applied technology lab of Institute of Plasma Physics Chinese Academy of Sciences as a master candidate. I am interesting in software engineering and Computer Sicence. Last year I noticed the recruit advertisement of iGem 2010 USTC_Software team in school BBS website. I make the decision to apply immediately. It's really happy for me during those past server months working with our team.
I play Ping Pong most during my spare time, because Ping Pong is a sport emphasizes on speed, skill and acting harmony ability rather than brute force. And I go jogging every day, which i think is a good way to relieve pressure and keep me in health!
I want to be a brilliant software engineer developing some brilliant works. I gain strong knowledge in software engineering and computer algorithms. IGEM 2010 is one step to my ambitions, it is a important project but not the last project of my engineering life, I wish I would develop more and more softwares in future. If you need help, do not hesitate to contact me please!
 Yubo Qi
Yubo QiI am Yubo Qi!
Yubo Qi 3005, national labotarory for physical sciences at the microscape
I am an undergraduate student in theoretical physics. But I am also interested in chemical physics and biology. In my labotarory, I concentrate on the calculation about transmission in nanostructure. However, biology science is also my favourite. So, I decide to join USTC_Software team of iGem 2010. Now I write database of biology reaction for my team. I love biology, chemistry, physics and decide to devote myself to the research of interdisipline in the future.
 Luojun Wang
Luojun WangLuojun Wang(Vic Wong)
Dept. of Statistics & Finance, USTC, Hefei, Anhui 230026, China;
Lab of Systems Biology of Hefei National Laboratory for Physical Sciences at the Microscale, USTC, Hefei, Anhui 230026, China
vicwong@mail.ustc.edu.cn; victorwangluo@gmail.com;
Hey, I'm Vic, an undergraduate senior student majoring in mathematical statistics. Yet my research field is biology statistics, and that is why I'm here.
For IGEM, I'm mainly responsible for human practice and have been giving lectures as well as other promotions on both synthesis biology and IGAM. It is this special experience that has not only made my college life amazing, but also presented me with a broader view of biology. Besides working for IGEM, I've also been researching biology statistics, including bioinformatics and biology networks.
My goal is to be a biostatistician in the future, on which I'm gonna keep putting effort. Now I'm applying for an opportunity to study as a biostatistics PhD. Good luck to me, huh? : )
 Jue Wang
Jue WangI am Jue Wang!:)
Dept. of Math, USTC, Hefei, Anhui 230026, China;
Email: juewang@mail.ustc.edu.cn
Hi,I am Jue, a Math majored senior student. As the boundaries among different disciplines of science are now blurring, I want to handle the ideology of maths to arrive at a higher standpoint to try to understand the principle of life. iGEM provides us with such a good state to learn from utilization.
In this group, I am the unique girl among nine boys, and the most frequently spoken of mine is "Oh, it's my duty to get some soy sauce! " Strange, huh? The connotation of it is not words but meanings. Those guys are too gentle to arrange me doing something of coding.So I am more involved in clerical works, I design the team uniform and our logo,arrange the scheduling and get in touch with sponsors and alumni connection. My life is full of unpredictable tasks, who can tell me what is the next?
 Zhaoyi Li
Zhaoyi LiMy profile:
Zhaoyi Li, Hefei National Laboratory for Physical Sciences at Microscale & National Synchrotron Radiation Laboratory, East Campus, USTC Hefei, Anhui 230026, China, lizhaoyi@mail.ustc.edu.cn </p>During my 2 years’ journey in the nano world, I got to know iGEM and joined in USTC Software team with great honor. Just as NEW YORK TIMES says, iGEM is a Do-It-Yourself Genetic Engineering Program. I am strongly attracted by this idea and cannot help imaging the science fiction picture where our daily life is changed by our genetic machine. It is unforgettable to have a brainstorm with talents from different academic background and turn our imagines into reality. Thanks to iGEM, I experienced a new trip in the synthetic biology and benefited a lot from my partners. Also, to compete against more than 1,000 other students from 100 schools, including many top-flight institutions is exciting.
I love traveling and always enjoy trips with my friends. I have a dream that one day I can travel around the world to discover the great scenery make new friends and experience different culture. Sports is also my favorite, I am good at playing football and gifted in table tennis.
 Zhenda He
Zhenda HeI am Zhenda he!
Hello , everyone! My name is He Zhengda , a student from Chemical Physics Department in University of Science and Technology of China . To be a member in 2010 USTC_Software is a really great time for me during the boring semester ! I met a lot of interesting people in the IGEM lab , and I wish to meet all you guys during the Jamboree . I have a enormorous interest in math , computer science and designing stuff . I am a Wiki interface designer in our team . So I hope everyone like it . It's a short description of me~ See you in the Jamboree!
 Shangyu Luo
Shangyu LuoI am Shangyu Luo!
Introduction: Hi all, I am Shangyu Luo. It is my Chinese name and my English name is Seraph. I am very pleased with attending iGEM competition with my teammates of USTC SOFTWARE. My major is computer science and I am interested in both computer and biology. During this competition, I gain a lot and make many friends. Thanks for my teammates, teachers and School of Computer Science and Technology! Additionally, I favor AI (Artificial Intelligence) in the fields of computer science, if you would like to create some beings together with me and to make them intelligent, please feel free to contact with me! Thanks all!
By Seragh
lsyurd@gmail.com
School of Computer Science and Technology
University of Science and Technology of China
- Instructors
-
Instructors
 Zhaofeng Luo
Zhaofeng Luo- Mail: lzf@ustc.edu.cn
 Hao Jiang
Hao Jiang- Mail:haojiang@mail.ustc.edu.cn
- Major: Cell Biology
- Research Interests: Synthetic Biology, Directed Evolution
- E-mail: haojiang@mail.ustc.edu.cn
- Former 2009 iGem USTC team member
Except where otherwise noted, content on this site is licensed under a Creative Commons Attribution 3.0 Licence.













































































