 "
"
Team:Lethbridge/Notebook/Lab Work/June
From 2010.igem.org
|
|
|
|
|---|

|

|

|

|

|

|

|

|

|

|
|---|
|
|
|

|

|
|
|---|

|

|

|

|

|

|

|
|---|---|---|---|---|---|---|
June 2010
June 1/2010
JV quantified the amount of DNA in gels run to date using ImageJ software. Results to be posted in working plasmids box.
Objective: Transform plasmids into DH5α
Method: Follow competent cell transformation protocol to transform the following:
From our ligations:
- pLacI-sRBS-Lumazine-dT
- pLacI-sRBS-Lumazine-dT
- mms6 (A6)
- mms6 (B6)
- xylE (C4)
- xylE (B4)
From the 2010 Parts Distribution:
- ECFP (Bba_E0020)
- EYFP (Bba_E0030)
- BglII Endonuclease (Bba_K112106)
June 2/2010
(In Lab: JV)
Objective: Isolate plasmid DNA of RBS-xylE (BBa_J33204) from DH5α cells and confirm results.
Method: "Mini-prep" the plasmid DNA using boiling lysis miniprep. Then restrict the DNA once and run on a 1% agarose gel (TAE).
Restriction Reaction
Ingredient Volume(µL) MilliQ H20 Water 15.75 Orange Buffer (10x) 2 pDNA (rbs-xylE) 2 EcoRI 0.25 Unrestricted Control
Ingredient Volume(µL) MilliQ H20 Water 16 Orange Buffer (10x) 2 pDNA (rbs-xylE) 2
DNA was restricted for 80 minutes at 37oC.
Analyzed results on a 1% agarose gel. Load order as follows:
Lane Sample Volume
Sample (µL)Volume Loading
Dye (µL)1 Restricted RBS-xylE 10 2 1 Unestricted RBS-xylE† 1 2 1 1kb Ladder†† 2 2 † Added 9µL MilliQ H2O
†† Added 8µL MilliQ H2O
Ran gel at 100V from 2 hours.
Results:
Conclusions: Plasmid DNA prep and restriction was successful.
Objective: Ligate rbs-xylE (Bba_J33204) to our double terminator, and insert it into the pSB1T3 plasmid backbone.
Method:
- Restrictions
Set up reactions as follows:
- Restrict rbs-xylE wit EcoRI and SpeI (Red Buffer)
- Restrict the double terminator with XbaI and PstI (Tango Buffer)
- Restrict pSB1T3 with EcoRI and PstI (Red Buffer)
Component Volume (µL) MilliQ H2O 15.5 Buffer 2 pDNA 2 Enzyme 0.25 + 0.25 Set up control reaction as follows:
- MilliQ H2O - 16µL
- Buffer - 2µL
- pDNA - 2µL
Incubated reactions for 65 minutes at 37oC
Killed enzymes by incubating reactions for 10 minutes at 65oC
- Ligation
Reaction set up as follows:Incubated reactions overnight at room temperature (total of 19.5 hours)
- T4 DNA ligase - 0.25µL
- rbs-xylE - 5µL
- dT - 3µL
- pSB1T3 - 8µL
- 10x Ligation Buffer - 2µL
- MilliQ H2O - 1.75µL
Killed enzymes by incubating reactions for 10 minutes at 80oC</ul>June 2/2010 - Evening
Objective: Set up new ligations of pLacI and sRBS-Lum-dT according to Tom Knight's protocol. Previous ligation had very little DNA.
Relevant Information:
- Want a final mass of 25ng of each pDNA in the ligation mix.
- Final concentration of pDNA in restriction digest should be 25-50ng/µL.
- Tom Knight's restriction reaction is 50µL, therefore there should be 1000ng pDNA in each restriction digest.
- Identified the following plasmids in our working plasmids box:
Common Name Location Concentration (ng/µL) Volume/rxn (µL) pLacI Maxiprep A9 990 ~1 pLacI (B1) A6 440 ~2 sRBS-Lum-dT (2) A1 965 ~1 sRBS-Lum-dT (1) A2 1145 ~1 sRBS-Lum-dT Maxiprep B8 4780 ~.2 sRBS-Lum-dT B7 4375 ~.25 sRBS-Lum-dT (1) G2 335 ~3 sRBS-Lum-dT (2) G3 965 ~2
- Make a 1:10 dilution of sRBS-Lum-dt maxiprep (D8) and sRBS-Lum-dT (B7). 0.5µL pDNA in 4.5µL water.
- Cut pLacI with EcoRI and SpeI
- Cut sRBS-Lum-dT with XbaI and PstI
- Cut pSB1T3 with EcoRI and PstI
- Will have total of 12 ligation reactions, want 12x2µL of pSB1T3 to add to each, therefore want 25µL of pSB1T3.
Method:
Restriction
Name [pDNA] (ng/µL) Volume
pDNA (µL)Volume
Water (µL)Volume
Buffer (µL)Enzymes Total Volume sRBS-Lum-dT (A1) 965 1 43.5 5 0.25µL XbaI
0.25µL PstI50 sRBS-Lum-dT (A2) 1145 1 43.5 5 0.25µL XbaI
0.25µL PstI50 pLacI Maxiprep (A1) 990 1 43.5 5 0.25µL EcoRI
0.25µL SpeI50 sRBS-Lum-dT Maxiprep(B8) 4780 2 (of 1:10 dilution) 42.5 5 0.25µL XbaI
0.25µL PstI50 sRBS-Lum-dT (B7) 4375 2.5 (of 1:10 dilution) 42 5 0.25µL XbaI
0.25µL PstI50 pLacI (D6) 440 2 42.5 5 0.25µL EcoRI
0.25µL SpeI50 sRBS-Lum-dT (G2) 335 3 41.5 5 0.25µL XbaI
0.25µL PstI50 sRBS-Lum-dT (G3) 540 2 42.5 5 0.25µL XbaI
0.25µL PstI50 pSB1T3 25 12.5 7 5 0.25µL EcoRI
0.25µL PstI50 Incubate for 30 minutes at 37oC (Start- 12:10pm; End- 12:40pm)
Heat kill enzymes at 80oC for 20 minutes
Ligation:
In a 10µL final volume, add:
- 2µL of sRBS-Lum-dT component
- 2µL of pLacI component
- 2µL of pSB1T3 component
- 1µL of T4 Buffer
- 0.25µL of T4 DNA Ligase
- 2.75µL of MilliQ H2O
Incubate for 30 minutes at room temperature to ligate
Incubate for 20 minutes at 80oC to heat kill
June 3/2010
Carried out protocol described in June 2/2010 - Evening
Analyzed results on 1% agarose gel.Load order as follows:
Lane Gel 1
SampleGel 1 Load Gel 2
SampleGel 2 Load 1 1kb Ladder 2µL dye, 2µL ladder
8µL MilliQ H2O1kb Ladder 2µL dye, 2µL ladder
8µL MilliQ H2O2 Restricted
sRBS-Lum-dT (A1)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(A9)+sRBS-Lum-dT(A1)10µL DNA
2µL Dye3 Unrestricted
sRBS-Lum-dT (A1)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(D6)+sRBS-Lum-dT(A1)10µL DNA
2µL Dye4 Restricted
sRBS-Lum-dT (A2)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(A9)+sRBS-Lum-dT(A2)10µL DNA
2µL Dye5 Unrestricted
sRBS-Lum-dT (A2)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(D6)+sRBS-Lum-dT(A2)10µL DNA
2µL Dye6 Restricted
sRBS-Lum-dT (B8)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(A9)+sRBS-Lum-dT(B7)10µL DNA
2µL Dye7 Unrestricted
sRBS-Lum-dT (B8)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(D6)+sRBS-Lum-dT(B7)10µL DNA
2µL Dye8 Restricted
sRBS-Lum-dT (B7)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(A9)+sRBS-Lum-dT(G2)10µL DNA
2µL Dye9 Unrestricted
sRBS-Lum-dT (B7)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(D6)+sRBS-Lum-dT(G2)10µL DNA
2µL Dye10 Restricted
sRBS-Lum-dT (G2)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(D6)+sRBS-Lum-dT(G3)10µL DNA
2µL Dye11 Unrestricted
sRBS-Lum-dT (G2)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(A9)+sRBS-Lum-dT(G3)10µL DNA
2µL Dye12 Restricted
sRBS-Lum-dT (G3)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(D6)+sRBS-Lum-dT(B8)10µL DNA
2µL Dye13 Unrestricted
sRBS-Lum-dT (G3)10µL DNA
2µL DyepSB1T3 Ligation of:
pLacI(A9)+sRBS-Lum-dT(B8)10µL DNA
2µL Dye14 Restricted
pLacI (A9)10µL DNA
2µL DyeRestricted
rbs-xylE10µL DNA
2µL Dye15 Unrestricted
pLacI (A9)10µL DNA
2µL DyeUnrestricted
rbs-xylE10µL DNA
2µL Dye16 Restricted
pLacI B1 (D6)10µL DNA
2µL DyeRestricted
pSB1T310µL DNA
2µL Dye17 Unrestricted
pLacI B1 (D6)10µL DNA
2µL DyeUnrestricted pSB1T3 10µL DNA
2µL Dye18 Unrestricted
dT10µL DNA
2µL DyepSB1T3 Ligation of:
rbs-xylE+dT10µL DNA
2µL Dye19 Restricted
dT10µL DNA
2µL DyeRan gel at 100V for 90 minutes.
Results:
Conclusions:
June 3/2010 - Evening
Objective: Repeat restriction of pSB1T3 and ligate with pLacI and sRBS-Lum-dT. Previous ligations all used up on gel.
Method:
Ligation:
In a 10µL final volume, add:
- 2µL of sRBS-Lum-dT component
- 2µL of pLacI component
- 2µL of pSB1T3 component
- 1µL of T4 Buffer
- 0.25µL of T4 DNA Ligase
- 2.75µL of MilliQ H2O
Incubate for 30 minutes at room temperature to ligate
Incubate for 20 minutes at 80oC to heat kill
Following ligation, transformed using transformation protocol. Plates incubated in 37oC incubator for 44 hours.
Results: Only plate pLacI (D6) + sRBS-Lum-dT (G2) + pSB1T3 grew; had 2 colonies. Control plate did not grow, acidentally plated on tetracycline plate instead of ampicillin (pUC19).
Follow-up: Inoculated 5mL LB media (tetracycline positive) with cells from the transformation plates and incubated at 37oC overnight. (June 5/2010).
Objective: Ligate pBad-TetR part with fluorescent protein part in pSB1C3 backbone.
Method:
Restriction
Name Volume
pDNA (µL)Volume
Water (µL)Volume
Buffer (µL)Enzymes Total Volume pSB-NEYFP (B4) .8 43.7 5 0.25µL XbaI
0.25µL PstI50 pSB-CEYFP (B5) .9 43.6 5 0.25µL XbaI
0.25µL PstI50 pBad-TetR) .3 44.2 5 0.25µL EcoRI
0.25µL SpeI50 NEYFP (E1) 4.3 40.7 5 0.25µL XbaI
0.25µL PstI50 NEYFP (E2) 0.3 42.2 5 0.25µL XbaI
0.25µL PstI50 Fusion CEYFP (E3) 3.9 40.6 5 0.25µL XbaI
0.25µL PstI50 Fusion CEYFP (E4) 2.0 42.5 5 0.25µL XbaI
0.25µL PstI50 Fusion CEYFP (E5) 3.0 41.5 5 0.25µL XbaI
0.25µL PstI50 CEYFP (E6) 0.6 43.9 5 0.25µL XbaI
0.25µL PstI50 CEYFP (E7) 0.5 44.0 5 0.25µL XbaI
0.25µL PstI50 pBad-TetR (F4) 2.5 42 5 0.25µL EcoRI
0.25µL SpeI50 pBad-TetR (F5) 1.7 42.8 5 0.25µL EcoRI
0.25µL SpeI50 pSB-CEYFP (G4) 2.9 41.6 5 0.25µL XbaI
0.25µL PstI50 pSB1C3 15.5 46 0.25µL EcoRI
0.25µL PstI62 Incubated at 37oC for 75 minutes.
- Used Red buffer for the EcoRI/SpeI and EcoRI/PstI digests
- Used Tango buffer for the XbaI/PstI digests
- Did not heat kill upon removal from incubation, put directly into -20oC fridge.
Continue Ligation on Saturday (See below).
June 5/2010
(In the lab:AS)
Objective: Ligate restriction products from June 3/2010.
Relevant information:
- Have 3 tubes of part 1 (pBad-TetR)
- In ampicillin backbone
- Have 10 tubes of part 2 (fluorescent protein - various)
- In ampicillin backbone
- Will have 30 combinations
- Will use pSB1C3 as plasmid backbone
- Used most of the pSB1T3 and want to save remainder for creating new backbone via PCR.
Method:
*Restriction digests were not heat killed after reactions. Freezing probably killed the restriction enzymes, but I will hea kill them at 80oC for 20 minutes anyways prior to adding Ligase.
- Cool on ice for 10 minutes before adding ligase.
Master Mix Volume/tube (µL) Total Volume (µL) DNA 6 --- 10x Buffer 1 32 T4 DNA Ligase .25 8 MilliQ H2O 2.75 88
- Add 4µL master mix to each DNA tube.
Follow-up: Ligation reactions will be transformed into DH5α cells
June 6/2010
(In Lab: JV, HS)
Objective:
Isolate the following plasmid DNA from DH5α:
- pLacI-sRBS-Lumazine-dT in pSB1T3 (colony 2)
- pLacI-sRBS-Lumazine-dT in pSB1T3 (colony 1)
Method:
Followed boiling lysis miniprep protocol. Eluted with 10µL Milli-Q H2O and RNase A.
Notes:
- Placed colony 2 in cell E10 of glycerol stocks and J6 of working plasmid box.
- Placed colony 2 in cell F1 of glycerol stocks and J5 of working plasmid box.
Objective:
Transformed the following plasmid DNA into DH5α cells:
- pBad-TetR-CEYFP: (F5+E6), (B10+E7), (B10+E6), (F4+E6), (F5+E4), (F4+E7)
- pBad-TetR-Fusion CEYFP: (F5+E3), (B10+E4), (F4+E3), (B10+E3), (B10+E5), (F4+E4), (F5+E4), (F5+E5), (F4+E5)
- pBad-TetR-pSB CEYFP: (F4+B5), (B10+G4), (F4+G4), (F4+B5), (F5+B5), (F5+G4)
- pBad-TetR-NEYFP: (F5+E1), (B10+E1), (F4+E2), (F5+E2), (B10+E2), (F4+E1)
- pBad-TetR-pSB NEYFP: (F5+B4), (F4+B4), (B10+B4)
- Positive control -> DH5α + pSB1C3
- Negative control -> DH%α + Milli-Q H2O
Method:
Followed Competent Cell Transformation protocol and used chloramphenicol as an antibiotic. We plated all 200µL of DNA onto the plates. The plates were incubated at 37oC from 4:30pm to 10:00am.Results:
None of the plates showed any growth.June 7/2010
(In Lab: JV, HB, TF, AV)
Objective:
Purification of DNA to increase ligation and transformation efficiency.
Method:
BioBasic Protocol for Purification of PCR products.
DNA (from Working Plasmid Box) Purified:
- pBAD-TetR (B10)
- pBAD- TetR (F4)
- pBAD-TetR (F5)
- EYFP (B1)
- pSB-NEYFP (B4)
- pSB-CEYFP (B5)
- NEYFP (E1)
- NEYFP (E2)
- Fusion CEYFP (E3)
- Fusion CEYFP (E4)
- Fusion CEYFP (E5)
- CEYFP (E6)
- CEYFP (E7)
- EYFP (E8)
- EYFP (E9)
- EYFP (E10)
- ECFP (F1)
- ECFP (F2)
- ECFP (F3)
- EYFP (G1)
- pSB-CEYFP (G4)
Objective:
Restrict and run 1% agarose gel of plasmids J5, J6, and pSB1T3 (June 2/10).
Method:Restrictions
Component Volume (µL) Restriction Enzyme (XbaI) 0.25 Buffer (Tango) 2 Plasmid DNA 2 MilliQ H2O 15.75 Incubated for 1 hour in 37oC incubator.
Heat shocked for 10 min at 65oC on heating block.
1% Agarose Gel
Lane Sample Load (µL) 1 1kb ladder 2 ladder + 2 dye (6X) + 8 MilliQ H2O 2 J5-pLacI-SRBS-Lumazine-dT(1) 10 DNA + 2 Dye (6X) 3 J5-pLacI-SRBS-Lumazine-dT(1)restricted 10 DNA + 2 Dye (6X) 4 J6-pLacI-SRBS-Lumazine-dT(2) 10 DNA + 2 Dye (6X) 5 J5-pLacI-SRBS-Lumazine-dT(2)restricted 10 DNA + 2 Dye (6X) 6 pSB1T3 10 DNA + 2 Dye (6X) 7 pSB1T3 restricted 10 DNA + 2 Dye (6X) 8 Empty 9 Empty 10 Empty Ran at 100V for 72 min.
IMAGE TO COME!!!!!!
June 7/2010 - Evening
(In Lab: AS, KG)
Objective:
Ligation of:
- pBAD-TetR (F5) + CEYFP (E6)
- pBAD-TetR (F5) + NEYFP (E1)
- pBAD-TetR (F5) + pSB-NEYFP (B4)
- pBAD-TetR (F5) + Fusion-CEYFP (E7)
- pBAD-TetR (F5) + Fusion-CEYFP (E3)
- pBAD-TetR (F5) + CEYFP (E7)
- pBAD-TetR (F5) + NEYFP (E2)
- pBAD-TetR (F5) + pSB-CEYFP (B5)
- pBAD-TetR (F5) + Fusion-CEYFP (E5)
- pBAD-TetR (F5) + pSB-CEYFP (G4)
- pBAD-TetR (F4) + CEYFP (E7)
- pBAD-TetR (F4) + NEYFP (E1)
- pBAD-TetR (F4) + pSB-NEYFP (B4)
- pBAD-TetR (F4) + pSB-CEYFP (B5)
- pBAD-TetR (F4) + CEYFP (E6)
- pBAD-TetR (F4) + Fusion-CEYFP (E4)
- pBAD-TetR (F4) + Fusion-CEYFP (E3)
- pBAD-TetR (F4) + pSB-CEYFP (G4)
- pBAD-TetR (F4) + Fusion-CEYFP (E5)
- pBAD-TetR (F4) + NEYFP (E2)
- pBAD-TetR (B10) + pSB-NEYFP (B4)
- pBAD-TetR (B10) + pSB-CEYFP (G4)
- pBAD-TetR (B10) + NEYFP (E1)
- pBAD-TetR (B10) + CEYFP (E6)
- pBAD-TetR (B10) + CEYFP (E7)
- pBAD-TetR (B10) + Fusion-CEYFP (E4)
- pBAD-TetR (B10) + pSB-CEYFP (B5)
- pBAD-TetR (B10) + Fusion-CEYFP (E5)
- pBAD-TetR (B10) + Fusion-CEYFP (E3)
- pBAD-TetR (B10) + NEYFP (E2)
Method:
FILL ME IN!!!!!!
June 8/2010
(In the lab: JV, AV)
Objective: Follow the overexpression of our pLacI-sRBS-Lum-dT construct.
Method: FILL ME OUT!!!!!!
Results:
Time OD600 F1 OD600 E10 0 0.118 0.103 30 0.133 0.111 30 0.145 0.116 90 120 0.160 0.124 150 0.120 0.093 180 0.129 0.100 210 0.145 0.122 240 0.158 0.145 270 0.171 0.178 300 0.194 0.222 330 0.223 0.280 360 0.252 0.364 390 0.296 0.458 420 0.338 0.557† 450 0.394 0.656 480 0.453 0.675 510 0.530 0.688 540 0.598† 0.706 600 0.633 0.752 660 0.653 720 0.679 ∞ 1.278 † Overexpression induced by adding 1mM IPTG.
Following overexpression, 1mL of cells was removed from the culture, spun down at ~13000xg for 20 seconds, excess media removed and rinsed with water.
Suspended cells in 8M urea, mixed with 6x dye and ran on 18% SDS-PAGE gel for 90 minutes at 200V. Gel stained overnight.
Results: IMAGE TO COME!!!!
Objective: Calculate quantity of DNA in pBad-TetR and fluorescent protein mini-preps by staining an agarose gel.
Method: Restrict plasmid DNA (done by AV,HB,TF on June 7/2010) and run on a 1% TAE agarose gel (JV).
Lane Gel 1
SampleGel 1 Load Gel 2
SampleGel 2 Load 1 1kb Ladder 2µL dye, 2µL ladder
8µL MilliQ H2O1kb Ladder 2µL dye, 2µL ladder
8µL MilliQ H2O2 Restricted
EYFP (B1)10µL DNA
2µL DyeRestricted
Fusion CEYFP (E3)10µL DNA
2µL Dye3 Unrestricted
EYFP (B1)10µL DNA
2µL DyeUnrestricted
Fusion CEYFP (E3)10µL DNA
2µL Dye4 Restricted
pSB-CEYFP (B5)10µL DNA
2µL DyeRestricted
Fusion CEYFP (E4)10µL DNA
2µL Dye5 Unrestricted
pSB-CEYFP (B5)10µL DNA
2µL DyeUnrestricted
Fusion CEYFP (E4)10µL DNA
2µL Dye6 Restricted
ECFP (F2)10µL DNA
2µL DyeRestricted
EYFP (E10)10µL DNA
2µL Dye7 Unrestricted
ECFP (F2)10µL DNA
2µL DyeUnrestricted
EYFP (E10)10µL DNA
2µL Dye8 Restricted
pSB-CEYFP (G4)10µL DNA
2µL DyeRestricted
pSB NEYFP (B4)10µL DNA
2µL Dye9 Unrestricted
pSB-CEYFP (G4)10µL DNA
2µL DyeUnrestricted
pSB NEYFP (B4)10µL DNA
2µL Dye10 Restricted
EYFP (G1)10µL DNA
2µL DyeRestricted
ECFP (F3)10µL DNA
2µL Dye11 Unrestricted
EYFP (G1)10µL DNA
2µL DyeUnrestricted
ECFP (F3)10µL DNA
2µL Dye12 Restricted
NEYFP (E2)10µL DNA
2µL DyepBad-TetR (F5) 10µL DNA
2µL Dye13 Unrestricted
NEYFP (E2)10µL DNA
2µL DyeRestricted
Fusion CEYFP (E5)10µL DNA
2µL Dye14 Restricted
pBad-TetR (B10)10µL DNA
2µL DyeUnrestricted
Fusion CEYFP (E5)10µL DNA
2µL Dye15 Unrestricted
pBad-TetR (B10)10µL DNA
2µL DyepBad-TetR (F4) 10µL DNA
2µL Dye16 Restricted
CEYFP (E6)10µL DNA
2µL DyeRestricted
pSB1T310µL DNA
2µL Dye17 Unrestricted
CEYFP (E6)10µL DNA
2µL DyeUnrestricted pSB1T3 10µL DNA
2µL Dye18 Restricted
NEYFP (E1)10µL DNA
2µL Dye10µL DNA
2µL Dye19 Unrestricted
NEYFP (E1)10µL DNA
2µL Dye20 Restricted
ECFP (F1)10µL DNA
2µL Dye21 Unrestricted
ECFP (F1)10µL DNA
2µL Dye22 Restricted
EYFP (E9)10µL DNA
2µL Dye23 Unrestricted
EYFP (E9)10µL DNA
2µL Dye24 Restricted
CEYFP (E7)10µL DNA
2µL Dye25 Unrestricted
CEYFP (E7)10µL DNA
2µL Dye26 Restricted
EYFP (E8)10µL DNA
2µL Dye27 Unrestricted
EYFP (E8)10µL DNA
2µL DyeRan gel at 100V for 90 minutes.
Results:
No bands visible except for pSB1T3 lanes, therefore could not quantify anything that had not already been quantified.
Also, purification of DNA done on June 7/2010 seemed to reduce amount of pDNA in sample.
Objective: Restrict mms6 (D9,D10) and dT (C1) so we can ligate the dT onto the mms6 coding region.
Method:
mms6 Restriction
- 2µL mms6 pDNA
- 2µL Red buffer
- 0.25µL EcoRI
- 0.25µL SpeI
- 15.5µL MilliQ H2O
dT Restriction
- 2µL dT pDNA
- 2µL Orange buffer
- 0.25µL EcoRI
- 0.25µL XbaI
- 15.5µL MilliQ H2O
Incubated for 1 hour at 37oC
Heat shock on heat block (80oC) for 20 minutes
Ligation
- 2µL dT pDNA
- 2µL mms6 pDNA
- 0.25µL T4 DNA Ligase
- 1µL 10x buffer
- 4.75µL MilliQ H2O
Analyze results on 1% TAE agarose gel
Lane Sample Load (µL) 1 1kb ladder 0.5 ladder; 2 dye; 9.5 MilliQ H2O 2 Unrestricted mms6 (D9) 10 DNA; 2 Dye 3 Restricted mms6 (D9) 10 DNA; 2 Dye 4 Unrestricted mms6 (D10) 10 DNA; 2 Dye 5 Restricted mms6 (D10) 10 DNA; 2 Dye 6 Unrestricted dT (C1) 10 DNA; 2 Dye 7 Restricted dT (C1) 10 DNA; 2 Dye 8 Empty 9 Empty 10 Empty Results:
Looks like the mms6 DNA was not cut at all. Therefore is doubtful that the ligations will work.
June 8/2010 - Evening
Objective: Transform pLacI-sRBS-Lum-dT constructs assembled via three antibiotic assembly on June 3/2010. Also transform mms6-dT constructs assembled today using old insertion method.
Method: Follow transformation protocol. Results: No colonies anywhere
June 9/2010
(in the lab: TF, JV)
Objective: Transform mms6-dT ligation reactions from June 8/2010.
Method: Followed transformation protocol and transformed the following:
- mms6 (D9) + dT (C1)
- mms6 (D10) + dT (C1)
- pUC19 (positive control)
- Water (negative control)
Results: No transformants on plates.
June 10/2010
(In the lab: AV, HB, JV)
Objective: Repeat ligation of mms6 (B9,D9,D10) and dT (C1).
Method:
mms6 Restriction
- 2µL mms6 pDNA
- 2µL Red buffer
- 0.25µL EcoRI
- 0.25µL SpeI
- 15.5µL MilliQ H2O
dT Restriction
- 2µL dT pDNA
- 2µL Orange buffer
- 0.25µL EcoRI
- 0.25µL XbaI
- 15.5µL MilliQ H2O
Incubated for 1 hour at 37oC.
Killed enzymes by heating to 80oC for 20 minutes
Ligation
- 5µL dT pDNA
- 5µL mms6 pDNA
- 0.5µL T4 DNA Ligase
- 2µL 10x buffer
- 7.5µL MilliQ H2O
Incubated at room temperature overnight.
Results:
Analyzed on 1% TAE agarose gel:
Lane Sample Load (µL) 1 1kb ladder 0.5 ladder; 2 dye; 9.5 MilliQ H2O 2 Unrestricted mms6 (B9) 5 DNA; 2 dye; 5 MilliQ H2O 3 Restricted mms6 (B9) 5 DNA; 2 dye; 5 MilliQ H2O 4 Unrestricted mms6 (D9) 5 DNA; 2 dye; 5 MilliQ H2O 5 Restricted mms6 (D9) 5 DNA; 2 dye; 5 MilliQ H2O 6 Unrestricted mms6 (D10) 5 DNA; 2 dye; 5 MilliQ H2O 7 Restricted mms6 (D10) 5 DNA; 2 dye; 5 MilliQ H2O 8 Unrestricted dT (C1) 5 DNA; 2 dye; 5 MilliQ H2O 9 Restricted dT (C1) 5 DNA; 2 dye; 5 MilliQ H2O 10 D9 + C1 Ligation (June 8/2010) 5 DNA; 2 dye; 5 MilliQ H2O Gel run for 80 minutes at 100V
Looks like everything was restricted. Ligations may work this time.
Follow-up: Ligation mixtures can be restriction tested and (if cutting works) transformed into DH5α cells.
Objective: Transform pDNA from distribution plates into DH5α cells to generate glycerol stocks for lab.
Method:
Remove DNA from the following locations on the 2010 distributions kits:
- double terminator (B0010-B0010; AKA B0017) from kit plate 2 well 6K (pSB1A2)
- double terminator (B0010-B0012; AKA B0015) from kit plate 1 well 23L (pSB1AK3)
Transform into DH5α cells using transformation protocol, with pUC19 as positive control (1ng/µL), and water as negative control.
Results:
- Positive control: TMTC (too many to count) colonies
- B0010-B0010: 41 colonies
- B0010-B0012: 120 colonies
June 14/2010 Evening
(In the lab: TF, DM, HS)
Objective: Restriction Digest of RBS-xylE with EcoRI and SpeI
Method:
Restriction Digestions
- Pipetted 15.5µL of water, into 2 reaction tubes (0.6mL) each.
- Pipetted 2µL of red buffer into each of them.
- Pipetted 2µL of rbs-xylE plasmid DNA into both tubes, but taking only the thawed liquid on the side.
- Because this was not proper protocol for aliquoting DNA, we thawed the DNA out completely and then put 2µL more of the rbs-xylE plasmid DNA in.
- Pipetted 0.25µL of EcoRI enzyme and 0.25µL of SpeI enzyme into the restriction reaction tube.
- Placed in incubator (37oC) for 1 hour.
- Placed in ice for 10 minutes.
Objective: Test Ligations of rbs-xylE with T4-Ligase from iGEM -20oC and from HJ's lab's -20oC.
Method:
- Pipette 12.75µL of Milli-Q H2O into 4 mincrocentrifuge tubes.
- Pipette 2µL of 10X T4-Ligase buffer into all 4 tubes.
- Pipette 5µL of the RD product into both iGEM ligase tubes and HJ-Lab Ligase tubes.
- Pipetted 5µL of the RD control into both iGEM ligase tubes and HJ-Lab ligase tubes.
- Incubate overnight
June 15/2010
(In the lab: JV)
Objective:Continue T4-Ligase check and confirm that it is functional.
Method:Run plasmid DNA;uncut, cut, and ligated on a 1% agarose gel (TAE)
Lane Sample Load (µL) 1 rbs-xylE 10 DNA solution + 2 Dye 2 rbs-xylE R.D. 10 DNA solution + 2 Dye2O 3 rbs-xylE [iGEM ligation] 10 DNA solution + 2 Dye 4 rbs-xylE [HJ ligation] 10 DNA solution + 2 Dye 5 rbs-xylE [iGEM ligation control] 10 DNA solution + 2 Dye 6 rbs-xylE [HJ ligation control] 10 DNA solution + 2 Dye 7 1kb ladder 2 ladder + 2 dye + 8 Milli-Q H2O 8 mms6 (B9) R.D. 10 DNA solution + 2 Dye 9 mms6 (B9) 10 DNA solution + 2 Dye 10 mms6 (D10) R.D. 10 DNA solution + 2 Dye 11 mms6 (D10) 10 DNA solution + 2 Dye 12 mms6 (D9) R.D. 10 DNA solution + 2 Dye 13 mms6 (D19) 10 DNA solution + 2 Dye 14 dt (C1) R.D. 10 DNA solution + 2 Dye 15 dt (C1) 10 DNA solution + 2 Dye
Result:Ran gel for 89 minutes at 100V. Gel was unreadable. I will redo the gel using and 8 well comb. IMAGE TO COME!!!!
Lane Sample Load (µL) 1 rbs-xylE R.D. 5 DNA solution + 1 Dye 2 rbs-xylE 5 DNA solution + 1 Dye 3 iGEM ligation 10 DNA solution + 2 Dye 4 iGEM ligation control 10 DNA solution + 2 Dye 5 HJ ligation 10 DNA solution + 2 Dye 6 HJ ligation control 10 DNA solution + 2 Dye 7 1kb ladder 2 ladder + 2 dye + 8 Milli-Q H2O Ran gel at 100V for 82 minutes.
Result:The gel "broke" while running. Jeff hypothesized the cause to be excessive heat. However, the DNA was still visible.IMAGE TO COME!!!!
Objective:Isolate plasmid DNA from DH5α cells.
Method:The plasmid DNA is:
- 1)double terminator B0017 (B0010-B0010) from 2010 kit plate 2:6K pSB1A2
- 2)double terminator B0015 (B0010-B0012) from 2010 kit plate 1:23L pSB1AK2
This will give us 2 additional dt's (3 total) into our working plasmid box to ligate onto the end of our constructs.
Used the boiling lysis miniprep protocol and elute with 50µL of Milli-Q H2O with RNAse A.
These plasmids (labeled 1 & 2 above) are in:
- working plasmid box: 1). J9 2). J10
- glycerol sotck 2010 box: 1). F2 2). F3
DNA was then purified using the protocol for purification of PCR products (BioBasic). DNA was eluted with 35µL of elution buffer.
June 15/2010 Evening
(In the lab: AS)
Adam: not convinced that the rbs-xylE was cut properly last night. There doesn't appear to be and band where the insert should be.
Objective: To confirm that EcoRI & SpeI are cutting (also PstI). Use rbs-xylE as test plasmid DNA.
Method: Digest rbs-xylE with the following enzyme combinations:
- EcoRI + SpeI (old [i.e. John's]) + Red Buffer
- EcoRI + SpeI (new) + Red Buffer
- EcoRI + PstI + Red Buffer
- EcoRi + Red Buffer
- SpeI (old) + Tango Buffer
- SpeI (new) + Tango Buffer
- PstI + Red Buffer
- No enzyme controls (Red & Tango Buffer)
Reaction mix as follows:
- Milli-Q H2O 15.5µL
- 10X Buffer 2µL
- pDNA 2µL
- Enzymes 0.25µL + 0.25µL
Incubated at 37oC for 1 hour . Analyze on 1% TAE Agarose gel as follows:
Lane Sample Load (µL) 1 No Enzyme Control (Tango) 10 DNA solution + 2 Dye 2 No Enzyme Control (Red) 10 DNA solution + 2 Dye 3 PstI 10 DNA solution + 2 Dye 4 SpeI (old) 10 DNA solution + 2 Dye 5 SpeI (new) 10 DNA solution + 2 Dye 6 EcoRI 10 DNA solution + 2 Dye 7 EcoRI + SpeI (old) 10 DNA solution + 2 Dye 8 EcoRI + SpeI (new) 10 DNA solution + 2 Dye 9 EcoRI + PstI 10 DNA solution + 2 Dye 10 1kb ladder 0.5 ladder + 2 dye + 9.5 Milli-Q H2Oe
Results: Everything cuts exactly as it should. Continue with ligation and analysis. IMAGE TO COME!!!!
June 16/2010
(in lab: JV) Objective: To miniprep Adam's overnight cultures of xylE and rbs. Completing this will give us a working stock of this plasmid.
Method: Use the boiling lysis miniprep protocol and pufrify using BioBasic protocol for purification of PCR products.
Objective: Transform pLacI-sRBS-lumazine synthase-dt into BL21(DE3).
Method:
- Followed transformation protocol
Changes to the protocol include:
- Added 5µL DNA to 50µL BL21(DE3).
- Incubated in 500µL SOC instead of 250µL LB
- incubated for 90 minutes at 37oC at 4:30pm.
Results: There wasn't any growth after overnight incubation. Possibility of an antibiotic mix-up.
June 16/2010 Evening
(in lab: ADS)
Objective: Continue with ligation test. Use restriction products to test T4 DNA ligase.
Method: Have 10µL of DNA remaining for each restriction. Ligate together one of the single cut reactions, and ligate one of the double cut reactions. 4µL of each sample will be tested against each ligase.
i.e.
- EcoRI +SpeI (new) cut vs HJ's ligase
- EcoRI + SpeI (new) cut vs iGEM ligase
- SpeI (new) cut vs HJ's ligase
- SpeI (new) cut vs iGEM ligase
Reaction Mixture:
- DNA -> 4µL
- 10X Buffer -> 2µL
- Milli-Q -> 13.5µL
- Ligase -> 0.5µL
Prior to setting up reaction, heat kill restriction enzymes by heating to 80oC for 20 minutes and subsequently cooling on ice for 10 minutes.
Ligations begun at 6:50pm
Will run samples at 30 minutes reaction time and overnight.
Analyze ligations in a 1% TAE Agarose gel
Lane Sample Load (µL) 1 No Enzyme Control (from last night) 1 DNA solution + 1 Dye + 4 H2O 2 EcoRI + SpeI (old)(from last night) 1 DNA solution + 1 Dye + 4 H2O 3 EcoRI + SpeI (new) vs HJ's Ligase 5 DNA solution + 1 Dye 4 EcoRI + SpeI (new) vs iGEM ligase 5 DNA solution + 1 Dye 5 SpeI (new) vs HJ's Ligase 5 DNA solution + 1 Dye 6 SpeI (new) vs iGEM ligase 5 DNA solution + 1 Dye 7 SpeI (old)(from last night) 1 DNA solution + 1 Dye + 4 H2O 8 1kb Ladder 0.25 Ladder + 4.75 H2O + 1 Dye
No observable ligation occurring 30 minutes with both ligases.
Objective: Prepare DNA for sequencing
Method: Need 20µL of DNA at 100ng/µL for each reaction. Need 20µL of 10µM primer for every 5 reactions.
Plasmids that need to be sequenced:
Name Concentration (ng/µL) Dilution Factor Final Concentration (ng/µL) A8-CFP complete 1395 1/5 280 B4-pSB NEYFP 1105 1/5 240 B5-pSB CEYFP 1055 1/5 210 B6-CFP complete 1285 1/5 260 D7-xylE 1820 1/10 182 D8-xylE 420 1/2 210 E1-NEYFP 235 1/2 117.5 E2-NEYFP 2980 1/10 300 E3-Fusion CEYFP 255 1/2 122.5 E4-Fusion CEYFP 490 1/2 245 E5-Fusion CEYFP 335 1/2 335 E6-CEYFP 1605 1/10 160 E7-CEYFP 1930 1/10 190 G4-pSB CEYFP 340 1/4 85 G5-CFP complete 355 1.4 89 Total of 15 primers -> 30 reactions
60µL of each VF2 & VR primers (10µL) -> send 65µLJune 17/2010
(in lab: JV, HB) Objective:Run Agarose gel of overnight samples from June 16/10 to test T4 DNA ligase
Method:Analyze ligation on a 1% TAE agarose gel.
Lane Sample Load (µL) 1 No Enzyme Control (from June 15) 2 DNA solution + 2 Dye + 8 H2O 2 EcoRI + SpeI (old)(from June 15) 2 DNA solution + 2 Dye + 8 H2O 3 EcoRI + SpeI (new) vs HJ's Ligase 10 DNA solution + 2 Dye 4 EcoRI + SpeI (new) vs iGEM ligase 10 DNA solution + 2 Dye 5 SpeI (new) vs HJ's Ligase 10 DNA solution + 2 Dye 6 SpeI (new) vs iGEM ligase 10 DNA solution + 2 Dye 7 SpeI (old)(from last night) 10 DNA solution + 2 Dye 8 1kb Ladder 0.5 Ladder + 9.5 H2O + 2 Dye Ran for 80 minutes at 100V.
Results:
IMAGE TO COME!!!!
June 17/2010 Evening
(in lab: JS, AS, KG)
Objective:Do PCR to observe ligations
Method:
Master Mix Volume/tube (µL) Total Volume (µL) MilliQ H2O 10.8 59.4 DNA 2 ------- 5X Phusion Buffer 4 22 10µM dNTP's .25 8 Forward Primer 1 5.5 Reverse Primer 1 5.5 Polymerase 0.2 1.1
Reaction Mixtures:
- HJ Double
- iGEM Double
- HJ Single
- iGEM Single
- controls (no forward or reverse primer and no DNA)
June 18/2010
(in lab: JV)
Objective:Observe the ligations from June 17/10 that were amplified using PCR.
Method:Observe using 1% TAE agarose gel electrophoresis. Ran the gel for 80 minutes at 100V.
Lane Sample Volume (µL) 8 1kb Ladder 2 Ladder + 8 H2O + 2 Dye 2 control no DNA 20 PCR reaction + 4 6X Dye and used 12 of this solution 3 control no reverse primer 20 PCR reaction + 4 6X Dye and used 12 of this solution 4 control no forward primer 20 PCR reaction + 4 6X Dye and used 12 of this solution 5 HJ single 20 PCR reaction + 4 6X Dye and used 12 of this solution 6 iGEM single 20 PCR reaction + 4 6X Dye and used 12 of this solution 7 HJ double 20 PCR reaction + 4 6X Dye and used 12 of this solution 8 HJ double 20 PCR reaction + 4 6X Dye and used 12 of this solution Results:IMAGE TO COME!!!!
June 21/2010
(in lab: JV)
Objective:Create more pSB1A3 plasmid.
Method:Run a PCR of the plasmid.
Master Mix Volume/tube (µL) MilliQ H2O 10.8 DNA 2 5X Phusion Buffer 4 10µM dNTP's .25 Forward Primer 1 DIlute 10X -> 0.1 + 0.9 MilliQ H2OReverse Primer 1 DIlute 10X -> 0.1 + 0.9 MilliQ H2OPolymerase 0.2 Use ligtest setting. WIll run 36 cycles.
Use 3 controls:
- no forward primer
- no reverse primer
- no DNA
Results:PCR samples were run on a gel (1% 1XTAE) with the PCR samples from June 22/10. This gel was run on June 22/10
June 22/2010
(in lab: JV, HS)
Objective:To overexpress pLacI-sRBS-lumazine synthase-dt in DH5α cells.
Method:Incubate cells in 500mL LB w/Tet. At OD 0.6 (600λ) induce overexpression of lumazine synthase with IPTG (1µM). Take To, T1, T2, T3 readings after induced with IPTG.
Time OD F1 (600λ) OD E10 (600λ) 0 0.078 0.072 60 0.122 0.110 90 0.128 0.125 120 0.140 0.138 150 0.149 0.157 180 0.169 0.183 210 0.205 0.230 240 0.243 0.276 270 0.278 0.323 300 0.317 0.398 330 0.394 0.428 360 0.394 0.480 390 0.441 0.553 400 ---- 0.594 (induced) 410 0.502 ----- 450 0.580 (induced) 0.736 510 0.804 1.005 570 1.065 1.066 630 1.053 1.043 ∞ 0.055 (1/10 dilution) 0.078 (1/10 dilution)
Objective:PCR amplify the following biobricks:
- dt (23L)
- dt (6K)
- dt (C1)
- mms6
- control (no DNA)
Method:
Master Mix Volume/tube (µL) Total Volume (µL) MilliQ H2O ---- 89.6 DNA 2 ------- 5X Phusion Buffer 4 24 10µM dNTP's .4 2.4 Forward Primer 0.2 1.2 Reverse Primer 0.2 1.2 Polymerase 0.2 1.2
1.2% Agarose gel electrophoresis (1X TAE), used to analyze the above PCR's. 25mL of agarose solution was used in the small gel rig.
Lane Sample Volume (µL) 1 pSB1A3 1 PCR sample + 2 Dye + 9 H2O 2 no DNA 1 PCR sample + 2 Dye + 9 H2O 3 no reverse primer 1 PCR sample + 2 Dye + 9 H2O 4 no forward primer 1 PCR sample + 2 Dye + 9 H2O 5 1kb ladder 0.5 ladder + 2 dye + 9.5 Milli-Q H2O 6 mms6 1 PCR sample + 2 Dye + 9 H2O 7 dt (23L) 1 PCR sample + 2 Dye + 9 H2O 8 dt (6K) 1 PCR sample + 2 Dye + 9 H2O 9 dt (C1) 1 PCR sample + 2 Dye + 9 H2O 10 no DNA 1 PCR sample + 2 Dye + 9 H2O
Ran the gel for 40 minutes at 100V.
June 22/2010 Evening
(in lab: KG, AS)
Objective:To run a PCR of:
- dt (J9,J10,C1)
- mms6 (B9,F10)
- rbs-xylE (I1,I2)
- xylE (D7, D8)
- pLacI-sRBS-lumazine synthase-dt (I8,I9)
- sRBS-lumazine synthase-dt (A1,A2,B7,B8,G2,G3)
- pLacI (A9,D1)
- pSB1A3
so that we have more DNA for restrictions and ligations.
Method:
Master Mix 1 Volume/tube (µL) Total Volume (µL) MilliQ H2O 10.8 226.8 DNA 2 4 5X Phusion HF Buffer 4 84 10µM dNTP's 1 21 Forward Primer (VF2) 1 21 Reverse Primer (VR) 1 21 Fimnzzymes polymerase 0.2 4.2
Master Mix 2 Volume/tube (µL) Total Volume (µL) MilliQ H2O 10.8 226.8 DNA 2 4 5X Econo Taq Buffer 4 84 10µM dNTP's 1 21 Forward Primer (VF2) 1 21 Reverse Primer (VR) 1 21 Imitrages polymerase (Econo Taq) 0.2 4.2
Master Mix for pSB1A3 Volume/tube (µL) Total Volume (µL) MilliQ H2O 10.8 31 DNA 1 20 5X Phusion Buffer 4 10 10µM dNTP's 1 2.5 Primer (SB-prep-2Ea) 1 2.5 Primer (SB-prep-3P) 1 2.5 Fimnzzymes polymerase 0.2 0.5
Master Mix for pSB1A3 Volume/tube (µL) MilliQ H2O 10.8 DNA 2 5X Econo Taq Buffer 4 10µM dNTP's 1 Primer (SB-prep-2Ea) 1 Primer (SB-prep-3P) 1 Imitrages polymerase 0.2
June 23/2010
(in lab: JV, HS)
Objective:: To run the pcr products on an agarose gel.
Method: Run on 1% agarose gel using the large gel rig.
Lane Sample Load (µL) 1 1kb DNA Ladder 0.5 Ladder + 2 Dye + 9 H2O 2 Phu-dt-J9 5 PCR sample + 2 Dye + 3 H2O 3 Phu-dt-J10 5 PCR sample + 2 Dye + 3 H2O 4 Phu-dt-C1 5 PCR sample + 2 Dye + 3 H2O 5 Fus-mms6-B9 5 PCR sample + 2 Dye + 3 H2O 6 Fus-mms6-F10 5 PCR sample + 2 Dye + 3 H2O 7 Fus-rbs-xylE-I1 5 PCR sample + 2 Dye + 3 H2O 8 Fus-rbs-xylE-I2 5 PCR sample + 2 Dye + 3 H2O 9 Fus-xylE-D7 5 PCR sample + 2 Dye + 3 H2O 10 Fus-xylE-D8 5 PCR sample + 2 Dye + 3 H2O 11 Fus-pLacI-sRBS-lum-dt I8 5 PCR sample + 2 Dye + 3 H2O 12 Fus-pLacI-sRBS-lum-dt I9 5 PCR sample + 2 Dye + 3 H2O 13 Fus-sRBS-lum-dt A1 5 PCR sample + 2 Dye + 3 H2O 14 Phu-sRBS-lum-dt A2 5 PCR sample + 2 Dye + 3 H2O 15 Phu-sRBS-lum-dt B7 5 PCR sample + 2 Dye + 3 H2O 16 Phu-sRBS-lum-dt B8 5 PCR sample + 2 Dye + 3 H2O 17 Phu-sRBS-lum-dt G2 5 PCR sample + 2 Dye + 3 H2O 18 Phu-sRBS-lum-dt G3 5 PCR sample + 2 Dye + 3 H2O 19 Phu-pLacI A9 5 PCR sample + 2 Dye + 3 H2O 20 Phu-pLacI D1 5 PCR sample + 2 Dye + 3 H2O 21 dt J9 5 PCR sample + 2 Dye + 3 H2O 22 dt J10 5 PCR sample + 2 Dye + 3 H2O 23 dt C1 5 PCR sample + 2 Dye + 3 H2O 24 mms6 B9 5 PCR sample + 2 Dye + 3 H2O 25 mms6 F10 5 PCR sample + 2 Dye + 3 H2O 26 rbs-xylE I1 5 PCR sample + 2 Dye + 3 H2O 27 rbs-xylE I2 5 PCR sample + 2 Dye + 3 H2O 28 xylE D7 5 PCR sample + 2 Dye + 3 H2O 29 xylE D8 5 PCR sample + 2 Dye + 3 H2O 30 pLacI-sRBS-lum-dt I8 5 PCR sample + 2 Dye + 3 H2O 31 pLacI-sRBS-lum-dt I9 5 PCR sample + 2 Dye + 3 H2O 32 sRBS-lum-dt A1 1 PCR sample + 2 Dye + 9 H2O 33 sRBS-lum-dt A2 1 PCR sample + 2 Dye + 9 H2O 34 sRBS-lum-dt B7 1 PCR sample + 2 Dye + 9 H2O 35 sRBS-lum-dt B8 1 PCR sample + 2 Dye + 9 H2O 36 sRBS-lum-dt G2 1 PCR sample + 2 Dye + 9 H2O 37 sRBS-lum-dt G3 1 PCR sample + 2 Dye + 9 H2O 38 sRBS-lum-dt A9 1 PCR sample + 2 Dye + 9 H2O 39 sRBS-lum-dt D1 0.5 ladder + 2 dye + 9.5 Milli-Q H2O 40 pSB1A3 control 5 PCR sample + 2 Dye + 3 H2O 41 pSB1A3 5 PCR sample + 2 Dye + 3 H2O 42 pSB1A3 control 5 PCR sample + 2 Dye + 3 H2O 43 pSB1A3 5 PCR sample + 2 Dye + 3 H2O
Ran gel for 45 minutes at 100V and stained in EtBr for 20 minutes and de-stained for 10 minutes.
- all Econe tubes are red
- all Phusion tubes are blue
Results: IMAGE!!!
Objective: Adam put in overnight cultures on June 22. I want to extract the plasmid DNA.
Method:
- Put in overnight cultures at 8:30pm and taken out at 9:30am next morning.
- All cultures were DH5α in LB w/ Amp.
- Cultures were incubated at 37oC.
- Next morning cultures were "minipreped" using boiling lysis miniprep protocol.
Results: Will view plasmid DNA extracted, on a 1% agarose gel (1X TAE) run at 100V for an hour.
Lane Sample Load (µL) 1 1kb Ladder 0.5 Ladder + 2 Dye (6X) + 9.5 Milli-Q H2O 2 CFP complete 2010 box C8 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 3 Fusion CEYFP 2007 box H5 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 4 NEYFP 2007 box J6 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 5 Fusion CEYFP 2007 box J5 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 6 CFP complete 2010 box A10 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 7 pSB-NEYFP 2010 box B6 (C1?) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 8 Fusion CEYFP 2007 box I5 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 9 xylE 2007 box B9 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 10 CEYFP 2007 box G6 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 11 CFP complete 2010 box A6 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 12 pSB-CEYFP 2010 box C5 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 13 pSB-NEYFP 2010 box B6 (C1?) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 14 pSB-CEYFP 2010 BOX C3 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 15 xylE 2007 box C4 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 16 CEYFP 2007 box H6 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 17 NEYFP 2007 box J6 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O
Picture to come!!!!!!!!!
June 24/2010
(in lab: JV, HS, AV)
Objective:: To run a PCR of biobrick parts from out working plasmid box.
Method:
Master Mix Volume/tube (µL) Total Volume (µL) MilliQ H2O 12.8 448 DNA 1 ------- 5X Phusion Buffer 4 140 10µM dNTP's 1 35 Forward Primer 0.5 17.5 Reverse Primer 0.5 17.5 Polymerase 0.2 7
Ran for 25 cycles on lig25 setting.
Objective:: Restriction Digest, Run on agarose gel, and ligate PCR products from June 23/10.
Method:
Six tubes for restriction digest:
Part 1 Part 2 Lane 8 (I2) Lane 4 (C1) Lane 19 (A9) Lane 8 (I2) Lane 19 (A9) Lane 8 (I2)
Restriction Digest Reaction Mixture:
Ingredient Volume (µL) Plasmid DNA 2 Enzyme 0.5 Buffer 2 Milli-Q H2O 15.5
Incubate at 37oC for 1 hour. Then heat kill the enzymes at 80oC for 20 minutes.
2% Agarose gel electrophoresis:
Lane Sample Load (µL) 1 50bp Ladder 1 Ladder + 2 Dye (6X) + 5 Milli-Q H2O 2 Fus-RBS-xylE (I2); Lane 8 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 3 Fus-RBS-xylE (I2) restricted; Lane 8 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 4 Phu-dT (C1); Lane 4 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 5 Phu-dT (C1) restricted; Lane 4 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 6 Phu-pLacI (A9); Lane 19(1) 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 7 Phu-pLacI (A9) restricted; Lane 19(1) 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 8 Phu-pLacI (A9); Lane 19(2) 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 9 Phu-pLacI (A9) restricted; Lane 19(2) 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 10 Fus-RBS-xylE (I2); Lane 8(1) 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 11 Fus-RBS-xylE (I2) restricted; Lane 8(1) 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 12 Fus-RBS-xylE (I2); Lane 8(2) 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O 13 Fus-RBS-xylE (I2) restricted; Lane 8(2) 5 PCR sample + 2 Dye (6X) + 5 Milli-Q H2O
Ran gel at 100V for 60 min and stained in EtBr for 15 min.
Picture to come!!!!!!!!!
Objective:: Run 1% agarose gel of PCR samples from June 24/10.
Lane Sample Load (µL) 1 A4-pBAD 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 2 A4-pBAD 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 3 A6-SRBS 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 4 A7-SRBS 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 5 A8-CFP Complete 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 6 A10-SRBS 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 7 B1-EYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 8 B2-N-term tag 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 9 B4-pSB-NEYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 10 B5-pSB-NEYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 11 B6-CFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 12 B10-pBAD-TetR 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 13 D3 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 14 D4-C-term tag 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 15 D5-C-term tag 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 16 D6-PLacI 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 17 E2-NEYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 18 E6-CEYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 19 E7-CEYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 20 1kb ladder 0.5 ladder + 2 Dye (6X) + 9.5 Milli-Q H2O 21 E8-EYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 22 E9-EYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 23 E10-ECFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 24 F1-ECFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 25 F2-ECFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 26 F3-ECFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 27 F4-pBAD-TetR 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 28 F5-pBAD-TetR 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 29 G1-EYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 30 G4-pSB-CEYFP 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 31 G6-pBAD(1) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 32 G7-pBAD(2) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 33 G8-N-term tag 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 33 G9-Lumazine 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 33 1kb ladder 0.5 ladder + 2 Dye (6X) + 9.5 Milli-Q H2O
Results:
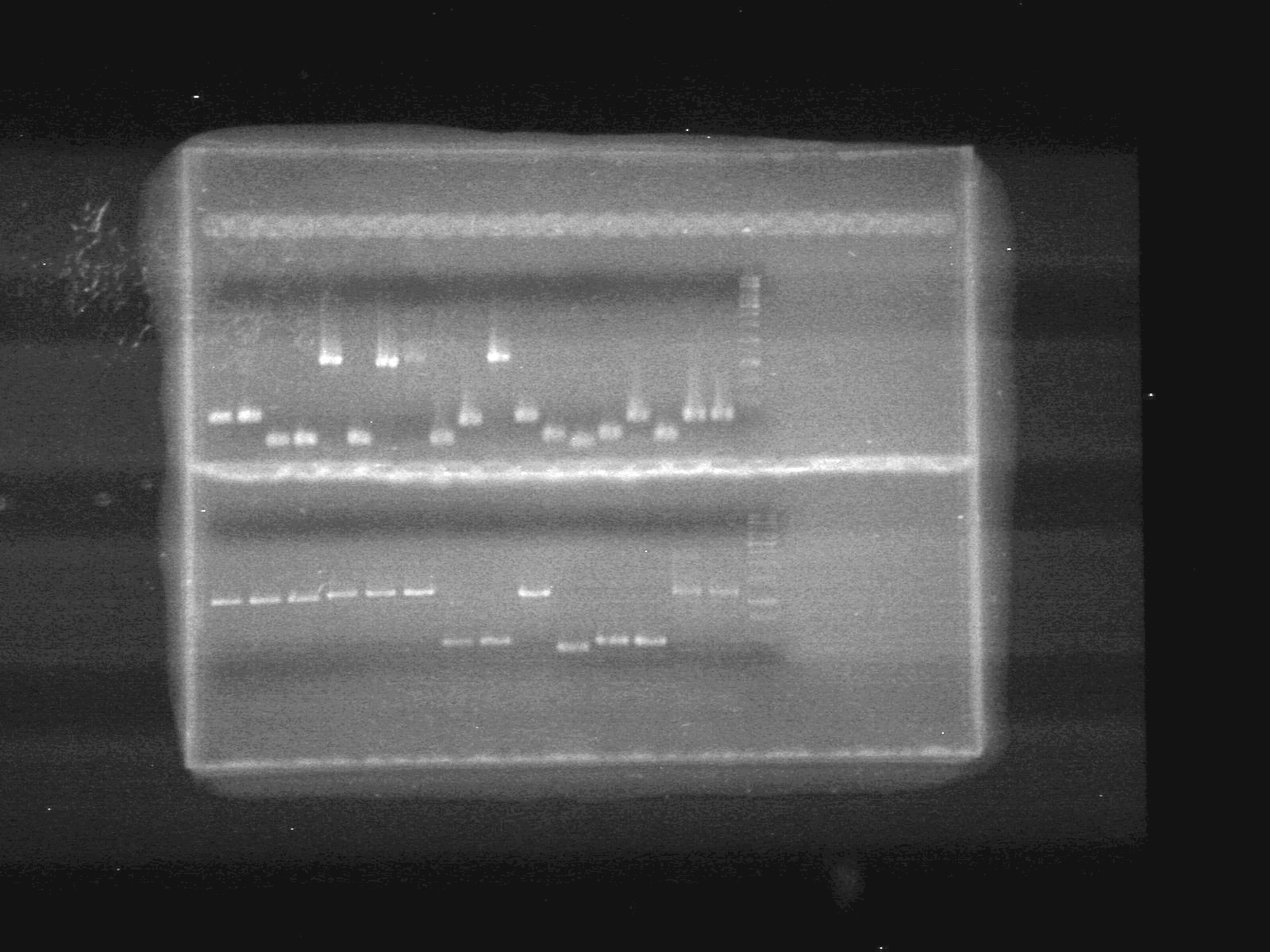
June 28/2010
(in lab: JV, HS, AV, HB)
Objective:: To restrict, run 1% agarose gel, and ligate all mms6, lumazine, xylE into pET28(a) for overexpression tests.
Method:
1) Restrict all parts and pET28(a) with NotI
2) Run on agarose gel (1%)
3) Ligate parts into pET28(a)
Parts:
- from working plasmid box 2010:
- A3-Lumazine
- B9-Mms6
- D7-xylE
- D8-xylE
- I10-Mms6
- from pink tray:
- B9-xylE
- C4-xylE
- G9-Lumazine
- (2X) pET28(a)
Restrictions:
- 2µL DNA
- 2µL Orange Buffer
- 0.25µL Enzyme (NotI)
- 15.75µL Milli-Q H2O
Protocol:
- 1) Incubate for 1 hour at 37oC
- 2) Heat kill for 20 minutes at 80oC (heat block)
Agarose Gel:
Lane Sample Load (µL) 1 1kb ladder 0.5 ladder + 2 Dye (6X) + 9.5 Milli-Q H2O 2 Lumazine A3 RD 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 3 Lumazine A3 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 4 Mms6 I10 RD 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 5 Mms6 I10 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 6 Mms6 B9 RD 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 7 Mms6 B9 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 8 xylE D8 RD 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 9 xylE D8 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 10 xylE D7 RD 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 11 xylE D7 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 12 Lumazine G9 RD (p) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 13 Lumazine G9 (p) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 14 xylE B9 RD (p) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 15 xylE B9 (p) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 16 xylE C4 RD (p) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 17 xylE C4 (p) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 18 pET28(a) RD 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 19 pET28(a) RD 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O 20 pET28(a) 5 DNA + 2 Dye (6X) + 5 Milli-Q H2O Ran gel at 100V for 90 minutes
Results:pET28(a) bands are all smeared, and are not useful from inserting parts.
June 30/2010
(in lab: JV)
Objective:: Isolate plasmid DNA from DH5α
Method:Used Kothe Maxiprep protocol.
Cell Pellet Weights:
- xylE = 1.15g
- Mms6 = 1.13g
- pET28(a) = 0.67g
- lumazine synthase = 1.1g
Results:



.jpg)


.JPG)