| When it comes to biobrick assembly, DNA of plasmid is often considered as an abstract straight line where biobricks can be placed on and connected together. It actually acts as the basic frame of biobrick assembly. Inspired by this, we present the abstract concept of chain, which also acts as the basic frame of part assembly. Chain is an abstract straight line where different parts can be placed on and connected together. Part is the basic unit of chain, and chain can have as many parts as possible.
|
| To go beyond DNA, and to unify the data structure of Species, we extend the concept of chain in MoDeL to include all the parts we can describe, including RNA, protein, and even compartment, compound and substituent, etc. Parts of DNA, RNA and protein, are all originated from DNA, and they are considered as “Sequence”. Sequence can be connected to other parts of sequence on a chain. On the contrary, parts of compartment and compound are considered as “Non-sequence”. Parts of non-sequence cannot be connected to any other part on a chain, which means, they can only exist on a One-Part-Chain. Substituent is used as a substitution of any other part, so it can be either sequence or non-sequence.
|
| It is important here to mention that the Sequence/Non-sequence division is only a rule guide the modeling. It is not a constraint to Chain-Node model. Actually, we choose to maximally preserve the flexibility and leave all the freedom of constructing a species to users. Users are allowed to construct ANY species they can using Chain-Node Model. For example, one can create a strange species with compartment, compound, and DNA parts on the same chain, which makes little sense in biology. So, it is really up to the users to decide how to use Chain-Node Model in modeling.
|
| Node is used to characterize binding structure in biological complex. Node is an abstract concept of binding site or site that can be bond. Links always exist between a binding site and the sites it binds together, indicating the relationship between them. We use trees to represent the binding structure in complex. The parent node represents the binding site, and the child node represents the bond sites. For example the binding structure of TetR dimer is considered as a TetR2 node with two TetR nodes as its children. There is one exception though, the ROOT node. ROOT node is used as the top node in a tree. It can only have one child and no parent. The function of ROOT is to indicate that its child is the last binding site in this tree, and it binds to nothing any more.
|
| In the reactions of promoter repression and activation, the promoter is repressed or activated after binding with repressor or inducer. So its properties are changed, and it is not the original promoter any more. This is an example where binding changes the properties of node. Since we do not store all the possible property alterations within one part, the solution is to replace the old part with a new part that stores the correct properties.
|
| Available nodes can be both a part on a chain or a node of binding site, which leaves a lot of possibilities in constructing complicated binding structure. Again, there are no restrictions, but parts of compartments are not suggested to appear in a binding structure.
|
| With great power there must come great responsibility. In order to use the powerful Chain-Node Model correctly, please have a look at a few examples we presents.
|
| Example of a species: E.coli cell
In MoDeL, the cell of E.coli is described as a species with only one part of E.coli(see Figure 1). Since the concept of part has been extended greatly, living cells can be described in the same way other species. Compartment belongs to “Non-sequence”, so it is suggested that E.coli only exists as a one-part-species.
|
 Figure 1: The Chain-Node Model of a E.coli cell |
| Example of a species: pLacI*:LacI4 and its tree structure of binding.
Here is another example of template species with more complicated structure.(see Figure 2) In biology, LacI tetramer (or LacI4) can bind with promoter pLacI as a repressor. When bond with LacI4, pLacI is repressed and its transcription speed will decrease significantly. In order to express this alteration, as mentioned above, we use another part of pLacI* with decreased transcription speed instead of pLacI in the species.
|
| The structure of binding sites is shown in figure (see Figure 3). The graph indicates that LacI monomers first bind together to form LacI dimer, then LacI dimers forms LacI tetramer, and finally LacI4 binds with pLacI* to form pLacI*:LacI4.
|
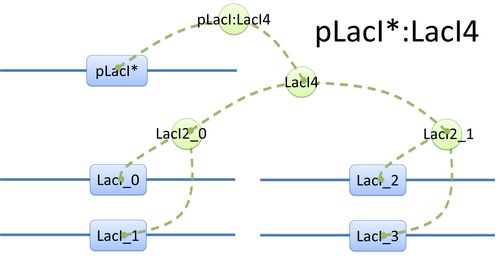 Figure 2: Species of pLacI*:LacI4 | 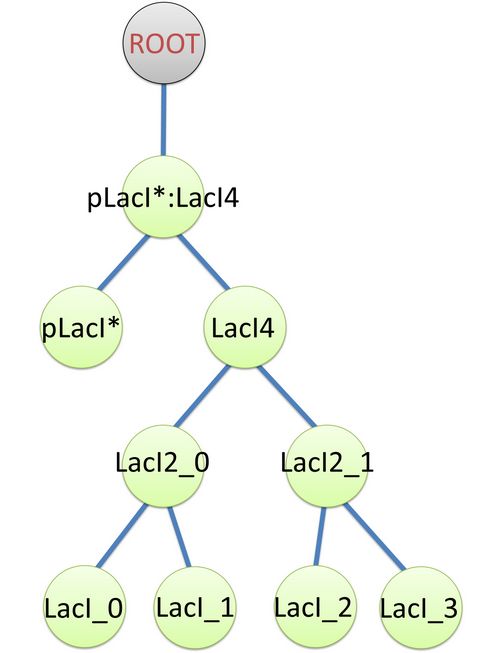 Figure 3: Tree structure of binding in pLacI*:LacI4 |
| There are two kinds of templates: Template Species and Template Reactions. They are designed to represent a family of species with similar structure and a family of reactions with similar mechanism respectively. In order to realize this idea, we add the container of Substituent into MoDeL. There are currently six objects in this container, ANY, ANYUB, ONE, ONEUB, NZ and NZUB. Though defined differently, they actually serve the same purpose: representing other parts. Let’s take ANY as an example (see Figure 4). ANY, represents parts on ONE chain and the number of represented parts can range from zero to infinity. For example, a DNA species with structure ANY - pTetR - ANY represents any DNA with part pTetR.
|
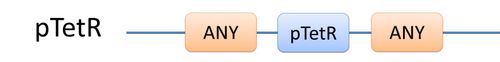 Figure 4: Template DNA species |
| A reaction with template species as its reactants, modifiers or products is a Template Reaction. A template reaction describes the core structure of reactant that causes the reaction to happen and provides a specification for generating reactions with the same mechanism. This could be understood more clearly by dividing parts in a species into functional groups -- a reaction template describes the interaction mechanism of these functional groups. For example, pTetR promoter is deactivated in presence of TetR dimer. The template species are pTetR DNA template and TetR dimer protein template (see Figure 5) and the functional groups are pTetR promoter and TetR dimer. All the parts of ANY should also be counted as a functional group, though they don’t have functions in this reaction. Any pair of species which partially contain pTetR DNA and TetR protein dimer respectively would bind according to the description of this reaction template. Modeling with templates allows users to define species and reactions only once for one certain family without rewriting them again in database.
|
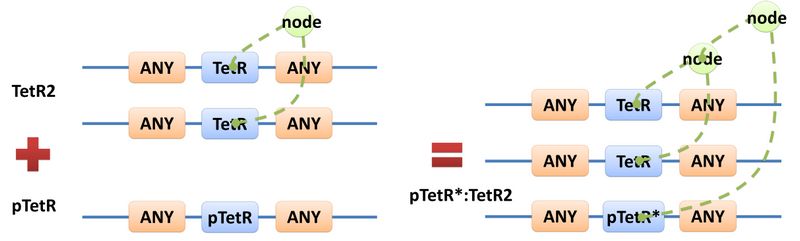 Figure 5: Template reaction |
| We are still looking for more substituents. We are planning to have more detailed substituents that can replace certain number of parts on certain number of chains. Even the arrangement of parts and chains can be specified. We believe it will make our template modeling more powerful.
|
| With plenty of information for automatic modeling needs to be stored, we design our database, and develop a standard language of describing the database. We call the language: MoDeL.
|
| MoDeL (Standard Biological Part Automatic Modeling Database Language) is a database representation format for synthetic biology automatic modeling. This project of MoDeL aims at providing a language and syntax standard for automatic modeling databases used in synthetic biology models on a number of topics, including cell signaling pathways, metabolic pathways, biochemical reactions, gene regulations, and so on.
|
| MoDeL is developed as a machine-readable format in XML fashion, programming languages with XML library API (such as libxml2 for C++) are strongly recommended for reading MoDeL database. USTC_Software has been devoted to the standardization of MoDeL. Documents can be found here. Example of database written in MoDeL can be downloaded on our [http://github.com/jkdirac/igame GitHub page].
|
| MoDeL is an attempt to define a universal database markup language for synthetic biology automatic modeling. It supports descriptions of a wide variety of different parts, including biobrick, compound, compartment, substituent, and so on. It can also describe the species constructed by these parts, and reactions happen with the species, such as binding of species, dilution of species, degradation of proteins, amplification of cells, multi-compartment reactions, and so on.
|
| Many traditional methods fail to consider structure of species when trying to markup reactions with only reactants, modifiers, products and kinetic laws. The best part of MoDeL is that it can preserve detailed structure in species and reactions using Chain-Node Model. Based on the model, MoDeL is able to markup almost all kinds of reactions in a detailed and clear way.
|
| Figure 5 above is an example of a simple network of biochemical reactions(pTetR + TetR2 = pTetR*:TetR2) that can be automatically constructed using MoDeL:
|
| MoDeL supports automatic construction of such network with only the input of its initial conditions and environmental parameters. In order to do this, obviously the data of parts, species, reactions, compartment, units and so on is needed. Besides, relationships between these components are also needed, so we can link to a reaction when we find a species. Data of relationship is stored as references and exists in most MoDeL components.
|
| To define all the needed components in a systematic way, MoDeL organizes them in five component containers at the top level:
|
| :: System: This container contains definitions of units, functions, rules and global parameters. These are all basic elements for other components of MoDeL, and many are mathematical concepts without direct synthetic biology meanings. The name of the container, System, separates this container from others which contains components of biological significance, and indicates this container includes the basic parameters of the whole system.
|
| :: Compartment: This container contains definitions of compartments. Compartment here is considered as a holder of substance with certain volume, so it only stores related data such as units and volume. It also stores a list of compartments that are permitted to be placed inside. For compartments like E.coli that can multiply, a corresponding compartment with the same id will be created in the sub-container of compartment in Part.
|
| :: Part: This container contains definitions of parts. Part includes the basic units that can be inserted in an abstract chain. The container of Part has five sub-containers which are: compartment, biobrick, compound, plasmidBackbone, and substituent. Biobrick contains biobricks from PartsRegistry together with other similar components created by users. Compartment contains cells and other microscopic organisms that can multiply. compound contains small molecules like IPTG and other compounds. PlasmidBackbone contains plasmid backbones come from PartsRegistry or defined by users. Substituent contains substitutes for template modeling.
|
| :: Species: This container contains definitions of various species defined by Chain-Node Model. Each component stores the cnModel structure of chains and trees. Related reactions are also stored. It is suggested that the constructor of a database stores template species instead of real species in this container. Only in this way can he make use of the power feature of template modeling of MoDeL.
|
| :: Reactions: This container contains definitions of various reactions. Each component has its own compartments, reactants, modifiers, products, substituent transfers, and kinetic law. This forms a detailed description of reactions. Again, constructor of a database is suggested to store template reactions instead of real reactions.
|
| The organizational structure of components in MoDeL is shown in the following figure.
|
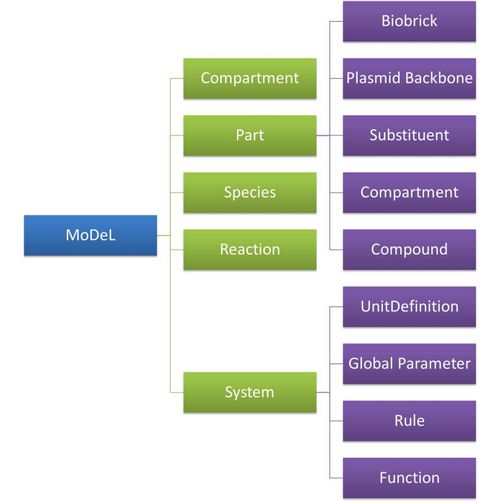 Figure 6: Structrue of MoDeL components |
| As mentioned above, reference relationships are stored in most components. The following figure shows the relationship between different components, the pointer pointing to a component indicates referencing to that component.
|
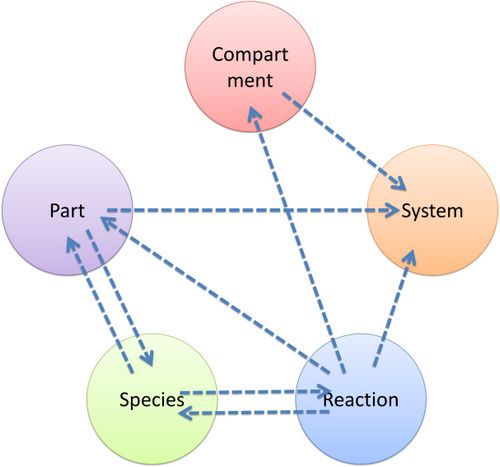 Figure 7: Reference relationship of MoDeL components |
| It is worth noting that MoDeL does not attempt to include all information about each component. In PartsRegistry, a long list of data in XML format is provided including part author, status, sequence, and even group access information. Such data are redundant for our database since they are not necessary elements needed to construct reaction network. Only the useful data is retained.
|
 "
"







