|
|
Experiment Design
In this section we do not only want to present the experiments and results we gained but also to encourage you to evaluate your own switch based on the the protocols and general prodecedure on how to evalutate basic parameters of a switch. In theory, every terminator can be turned into a switch with minor modifications and the right signals which are based on individual applications. While the principle of how to turn a terminator into a switch is explained in detail here, experimental setups and protocols are provided in the following. Due to time and equippment limitations we could not perform all the experiments we planned but next to the hope that another iGEM team might proceed with our project we would also like to encourage you to design and test some basic switches on which you can base a complete, tightly regulated network.
Read more
The complexity of our experimental setups vary, since we planned to characterize an individual switch with one exemplary signal on all relevant levels: Starting from the most general, complicated but also relevant level, in vivo measurements we approached to testing different switches on each smaller scale: We developed setups for in vitro translation which can be done without much effort following the in vitro measurements and also provide detailed description of in vitro transcription verification providing an inside to the molecular functionality of our basic idea. We do not see the methods we used here as the gold standart for bioLOGICS evaluation and encourage you to include your own ideas as well as check in our outlook section where we suggest experiments we could not do during the limited iGEM 2010 time. Together with our Biobrick submissions this year, we offer a complete set for switch evaluation on all cellular levels.
Most measurements are based on fluorescence reporters which provide easy handling, fast output and are well studied. Next to the fluorescent proteins GFP and mCherry we used in vivo, a malachite green binding aptamer serves as a reporter in vitro providing a reliable fluorescent output upon antitermination.
Most setups up to now were only used to evaluate switches with an default state "off" which are applied for AND/OR devices. In principle the same methods can be used for NOT devices which are based on a switch with an default state "off". Again, time limitation circumvented further tested from our team but we hope that further studies can be done in the future.
Close
In vivo Measurements
In vivo measurements have the highest complexity compared to any other experimental set-up. Different parameters and circumstances deriving from both the cellular environment as well as technical considerations like scatter have to be taken into account. Nevertheless, the measurements are essential, as our switches should finally work inside cells to fulfill our vision of an intracellular logic network. This year's submitted Biobricks provide you with a basic kit of plasmids which allow a quick beginning of the measurements.
Read more
Design
For the measurements in vivo we decided to use an expression cassette consisting of Green Fluorescent Protein (GFP) coding sequence upstream of the switch and another fluorescent protein coding sequence downstream of it. Both protein coding sequence share the same ribosome binding site allowing the usage of the GFP as an internal control in measurements. Since the spectra should not overlap and to avoid FRET as well as an pure overlap of the spectra, we settled on the usage of red fluorescent protein variants, namely mRFP1 in the first try and mCherry in an modified variant of the pSB1A10 vector.
While the GFP fluorescence can be used to normalize the measurements, the RFP fluorescence serves as a reporter to detect and evaluate termination and antitermination. To stimulate the expression of the fluorescent proteins, we took advantage of the pBAD promoter family (sensitive towards arabinose). The signal upon which the antitermination events and therefore switching relies where under the control of an IPTG inducible promter. We went with this well-established pair of controlable promoters to deliver an easy setup in the beginning, like described here, every sort of input may later be combined with our basic switching units.
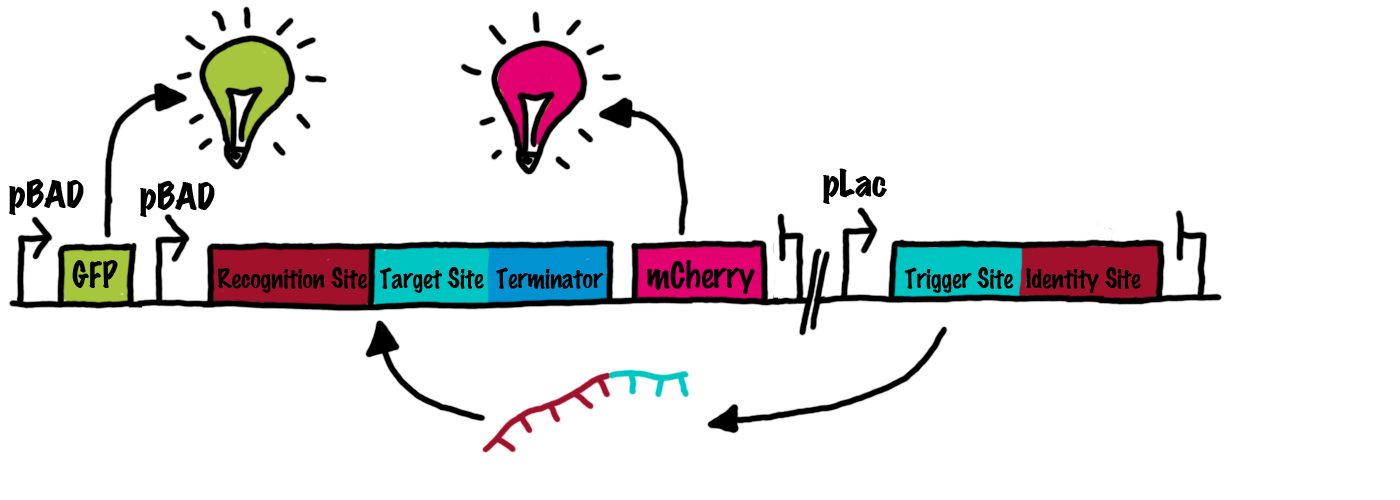 general measurement principle
The GFP internal control carries the advantage that errors in the measurement set can be detected easily. Lack of arabinose or promoter insensitivity can be recognized as well as problems with the fluorescence measurement itself. Plus, it allows normalizing measurements to compare different preparations in relation to each other.
Upon binding of a signal to the terminator switch, termination is circumvented and the reporter protein behind the switch can be translated. In the experimental setup presented here, this will result in an RFP expression, but again, every protein or DNA-encoded element in general may be used as an output. Since the RFP fluorescence spectra does not overlap with GFP if offers an easy possibility to evaluate the effect of signal induction. Next to GFP fluorescence, RFP fluorescence will show up.
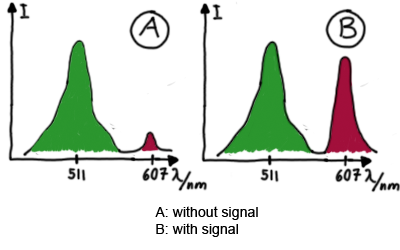 schematic estimated fluorescence spectra
Construction and Cloning
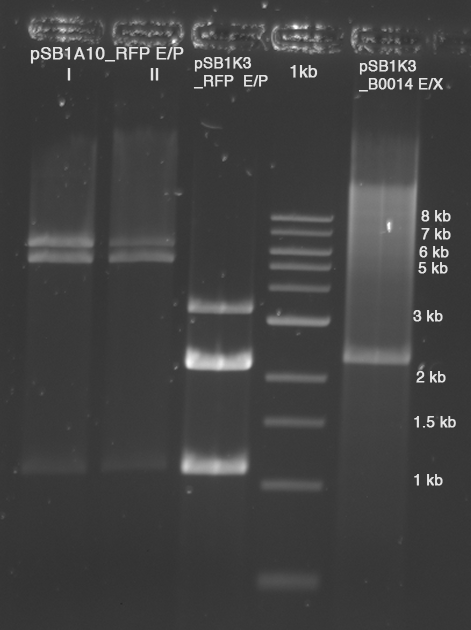
In a first try we cloned a measuring contruct based on pSB1A10. The resulting plasmid, nicknamed pMonsterplasmid due to its size was tested in the fluorescent measurements described below. Unfortunately after two months of cloning we had to recognize that the plasmid in use did not work for us (see also pSB1A10 Falsification).
So after the first unsuccessful attempts we decided to reclone the system, substituing RFP to mCherry, a dsRED derivative with a spectrum in the far red, and adding arabinose inducible promoters in front of both fluorescent proteins to guarantee stable and comparable expression of both proteins
To control the expression of the switch, the particular DNA sequence itself is under the control of an IPTG dependent promoter. In the future we want our networks to be able to respond to a variety of external signals like small metabolites, ions or whatever can be found in the parts registry. For basic switch evaluation, an established and well-working system like the lac-operon was chosen to avoid side-effects of less well-characterized promoters.
Measurements based on submitted Biobricks
The Biobricks BBa_K494001-BBa_K494006 are constructed for easy design of a switch-evaluation system. Detailed information can be found here.
Switch evaluation in vivo
To evaluate the switching efficiency, output with and without signal needs to be monitored. In this case, GFP fluroescence (internal control) will always appear upon arabinose induction, while RFP/mCherry fluorescence is only present upon binding of a signal and occuring antitermination.
Upon induction with arabinose a rise of GFP expression can be seen. To monitor changes in gene expression we used a fluorimeter and measured fluorescence of whole living cells. While this approach provides easy handling and monitoring, too much scattering has to be carefully avoided: the cell density should not exceed an OD600 of 0.05. RFP/mCherry emission should be visible only in case of a working switch or inefficient terminatrion.
For evaluation of the measuring plasmid itself we incorporated a positive control in every measurement. A random sequence in between GFP and RFP/mCherry was chosen in a corresponding length instead of a terminator. An increase in both GFP and mCherry was detectable in comparable amounts after quantum yield correction, showing the measuring plasmid to beworking nicely. While the positive control may be the same for all evaluated devices, the negative control has to be specific for every switch and terminator, respectively.
 Bacterial cultures after incubation of 16h
The negative control contained the evaluated switch without any possibility for induction of the corresponding signal. Thereby the switch's function is limited to termination, leading to no detectable RFP/mCherry fluorescence. By definition every switch type has to be tested using a negative control without a correponding signal, since termination efficiency may vary depending on the terminator itself, cell strain and general growth conditions. We recommend to chose your terminator of choice and evaluate it using the provided plasmids.
In our experimental part we evalutated terminators based on the regulatory unit of the tryptophan (Trp-Term) and histidine (His-Term) operons. Those synthetic operons are regulated based on the principle of attenuation, a terminator in front of genes involved in amino acid biosynthesis avoids transcription until environmental stimulis suggest a lack of those amino acids. Since both sequences are known to be regulated by changes in secondary structure, those two attenuators became the basis for our designed switches.
The terminators we tested can be found in the partsregistry. With the construction of the backbone [http://partsregistry.org/Part:BBa_K494001 BBa_K494001], potential switches and signals can be easily subcloned in two steps and tested. [http://partsregistry.org/Part:BBa_K494002 BBa_K494002] was constructed as a positive control, producing maximal mCherry fluorescence which may be used to characteize terminator and switch efficiency. [http://partsregistry.org/Part:BBa_K494003 BBa_K494003] and [http://partsregistry.org/Part:BBa_K494004 BBa_K494004] carry the His-Terminator with and without the corresponding signal, [http://partsregistry.org/Part:BBa_K494005 BBa_K494005] and [http://partsregistry.org/Part:BBa_K494006 BBa_K494006] being the same for Trp-Terminator.
Close
In vitro Translation
To go more into detail, the next complexity level is to study the effect of switches on a translational level. In vitro measurements with E. coli lysate make the fluorescence signals independent of cell growth and physical or biological factors like cell density or growth stadium.
Read more
Design
Since the same constructs can be used both for in vivo and in vitro translation, no additional cloning effort is needed. This implements, that the Biobricks we provided this year, can again be used as the groundwork for constructing vectors for measurements.
Reporter proteins GFP and mCherry are well expressed in vitro, the limiting factors are mostly the capacity of a kit versus the maturation time of fluorescent proteins. Since we used a fast-folding GFP variant and mCherry, which was characterized with a maturation time of 15 minutes by Tsien and Coworkers, the problem should be minimized. Alternative tags may be considered, a major advantage of measuring translation in vitro may be the use of non-cell permeable tags for switch evaluation.
Measurements
We used the cell-free E. coli S30 extract system for circular DNA provided by Promega[1], which is prepared by modifications of the Method Zubay et al.[2]. The characterization of the kit can be obtained from the [http://partsregistry.org/Cell-free_chassis/Commercial_E._coli_S30 Parts Registry].
Experiments were performed at 37°C with an amount of approximately 1 µg plasmid in a reaction volume of 50 µL. Fluorescence was followed over time in a jasco fluorolog with wavelength corresponding to those used in vivo.
Close
In vitro Transcription
To monitor transcription termination and antitermination on a the molecular level, in vitro transcription of individual switches and their response to signals offer an elegant way for fast and easy prove of principle. Most side effects occuring in a complex environment given in a cell or a cell lysate do not arise here. Another major advantage of in vitro transcription experiments is the possibility to test many signals for one switch to optimize antitermination efficiency and binding specifity without much cloning work. Data gained by in vitro transcription experiments can be used to improve switches and signals for in vivo usage.
Read more
Since we are working on a totally new principle of trancriptional control, we used this approach beside the above mentioned advantages for easy variation of different variables like the length of the core unit and the switch to signal ratio.
To study the switches on a transcriptional level offers the advantage, to reduce interference and possible artefacts to a minimum. Since we are not sure how cellular mechanisms like degradation of RNases or interacting factors as well as molecular crowding influence our systems, in vitro transcription was also used as a minumum system from which more complexity can derive.
Working with in vitro systems also has the advantage that an input is not needed anymore and the output can also be generated easily. We used two readouts with two different transcription systems to check and investigate our devices: First, we used an malachitegreen-binding aptamer for an fluorescence output and second, we simply put our reaction educts on an denaturing acrylamide-gel to check for RNA varying in length. As for two different transcription systems we used on the one hand E. coli-RNA Polymerase (RPO) based transcription since the aim is to apply the so gained results in vivo and on the other hand T7 based transcription which is well established through literature and delivers good RNA yields.
T7 RNA polymerase
The T7 RNA polymerase is known for satisfying RNA yields together with easy handling. In our approach we had PCR amplified, double stranded switches with an malachitegreen binding aptamer following after a T7 terminator which was constructed to function as a switch. Different signals were tested varying in length of the specifity site and the triggering unit.
For in vitro expression the T7 RNA Polymerase requires a double stranded promotor region at the beginning of the DNA template but is otherwise capable of handling single stranded DNA, so a sense strain corresponding to the T7 promoter region was added. Transcription is more effective with double stranded DNA as template. Apart from that, no more requirements are needed in theory which makes the evaluation of many signals especially easy. Since we ordered the signal sequences we tested we chose the cheaper way in the beginning by using single stranded signals with corresponding sense T7 pieces and switched to double stranded constructs after narrowing down the most promising switch/signal pairs. Later on we also used double stranded signals and switches since transcription rates are higher with those.
As a positive control, the malachite green binding aptamer right behind the T7 promoter was used. Transcription proceeds without termination and the maximal fluorescence intensity should be gained.
Transcription termination can also be estimated by measuring just the switch without interfering signals. Since upon transcription of a signal sequence, less RNA Polymerase is available, the transcription rate of the switch and therefore the fluorescence output is reduced by merely adding the signal. Therefore randomly chosen short sequences in the range of the tested signals were added to the negative control.
E. coli RNA polymerase
In comparison to the T7 RNA Polymerase the E. coli RNA Polymerase requires slightly more sophisticated proceedings when it comes to the design of switches and handling of the enzyme. The biggest in our case was to store it properly since the only -80°C fridge was in another building, so make sure you have a big supply of dry ice ready if you encounter the same problem.
E. coli RPO was ordered saturated with σ70-factor. The investigated terminators of an ???-promoter, the switch itself followed by a malachite green binding aptamer coding sequence.
Denaturing Polyacrylamide gel electrophoresis
Polyacrylamide gel electrophoresis (PAGE) was used for evaluation of termination and switching efficiency. Gels containing 15 % acrylamide and 6 M urea were used for separation of terminated and readthrough RNAs. The same constructs as designed for the malachite green binding aptamer were used.
Polyacrylamide gels seperate RNA and DNA according to their size in an electric field. Since the negative charge equals the size of nucelotides in the RNA/DNA, the number of base pairs can be compared between two samples often with one base pair resolution. Since RNA forms three-dimensional structures, the samples are preheated and run in 6 or 7 M urea. The polyacrylamide gel is stained in SybrGold afterwards which binds to both single and double stranded DNA and RNA. A Dnase digestion was applied before running the samples to avoid confusion caused by DNA templates.
Denaturing PAGE is a simple yet elegant way to check for transcription efficiency and termination rates. Since it is a very direct way and it provides a simple yet clear readout, we used it as another method beside the more sophisticated malachitegreen binding assay to evaluate and characterize our switch. Equippement for denaturing PAGE can be found in nearly every biochemical lab, so this method also applies for an easy controlexperiment.
Malachite green assay
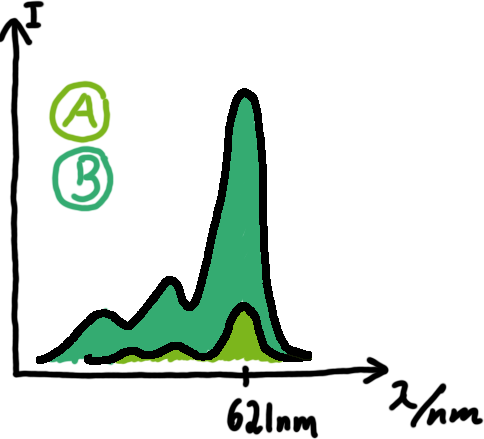 Emission spectra of malachite green; A: without signal-RNA, B: with signal-RNA In this year's DNA submission we contribute the malachite green binding aptamer which can be used as a transcription reporter in in vitro transcription experiments.
Malachite green is a dye with a negligible fluorescence in solution but undergoes a dramatic increase about 3000 times if bound by the RNA aptamer making it an exceptional good marker. Since the binding is very specific, transcription in dependence of a signal can be monitored by measuring the fluorescence of malachite-green over time if the aptamer is located behind the switch. Transcription of the aptamer will only take place after anti-termination by a signal. An increase should be visible over time. Other triphenyldyes are also recognized with weaker effects on the fluorescence but may also serve as reporters if the emission or excitation of malachite green does not fit the experimental setup.
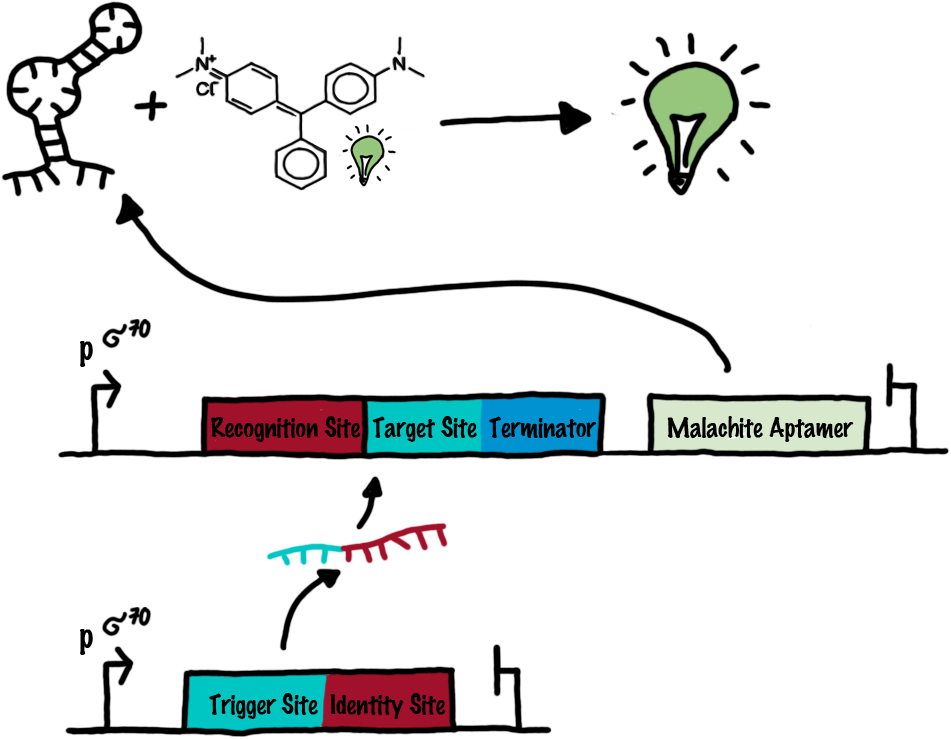 Description of the malachit green assay. Antitermination allows the polymerase to produce the malachite green aptamer Malachite green binding can be used to follow RNA transcription over time, a rise in the fluorescence is then detectable. Fluorescence marker of specific RNA structures are still rare, so the malachite green binding aptamer provides one of the only possibilities to continuously monitor transcription reactions. In comparison to PAGE, kinetics can be taken, while with PAGE only end point estimations can be made. This makes the malachite green binding aptamer a valuable tool to study in vitro transcription in general and the principles underlying our switch in principle.
Close
Lab Book
Explanations
In the following we present an overview regarding our work in the lab. For easier understanding we summarized the work of each week using colored boxes. To get a better overview we used the following color code for the boxes:
| The red box represents general cloning steps that were required for our measurements. See the protocol section for further details.
|
| The blue box indicates in vivo measurements which are described here.
|
| The green box indicates in vitro measurements relying on in vitro transcription and malachite green measurements. Details can be found here.
|
| The yellow box represents measurements done with an in vitro translation kit and is described in more details here.
|
To learn more about the work and results of a specific week, just click on the according week number. You will find detailed notes on our daily lab work. We present these notes in an unedited form as a record of our work, for for processed results please check the results section on our project page.
Chronological Lab Book
Week01
in vivo constructs
08.04.2010
Flo & Philipp
PCR
- samples:
- R0011_His
- R0011_Trp
- Control
- protocol: protocol
- templates: purified PCR products from 5.2.2010
- primer G1004/1005
- polymerase: Taq
- programm: igempcr
Purification of PCR products with QIAquick PCR purification Kit
- protocol followed. exceptions: DNA-binding/unbinding with 3min 6000rpm followed by 60sec full speed
2% Agarose Gel
09.04.2010
Philipp & Flo
Gel of PCR products from 08.04.2010
- loaded: 10 µL sample+2 µL 6x GLD, 4/2 µL LMW standard
- 110 V, 90 min
- stained with Sybrgold, 20 min, 1:10.000 dilution in TAE
- Standard - Control - R0011_His - R0011_Trp - Standard(=low molecular weight (see Protocols#Lab_Protocols))
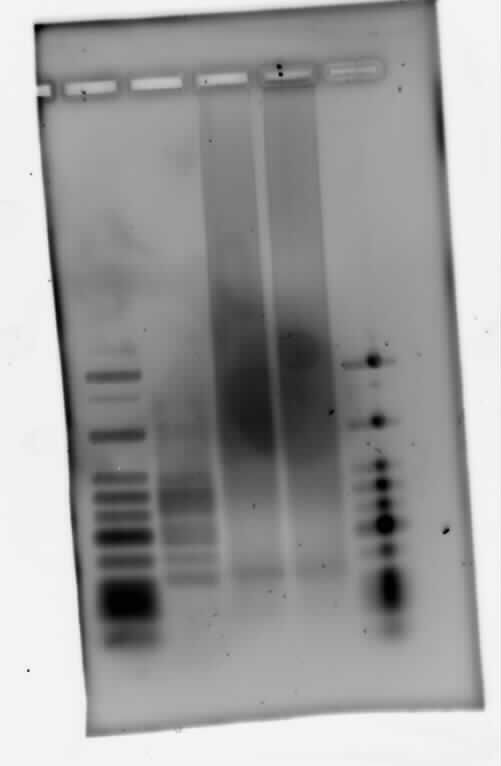
Close
Week02
in vivo constructs
15.04.2010
Philipp & Flo
[http://web.e14.physik.tu-muenchen.de/igem/index.php/Protocols#PCR PCR] of B0014 and R0011
16.04.2010
Philipp & Flo
- Concentrations measured with nanodrop:
| B0014
| 2.5 ng/µL
|
R0011
| 27.5 ng/µL
|
--> worked for R0011, not for B0014
- PCR of B0014
- Purification with the Zymo Kit, Elution in 20 µL H2O
- Concentration measured with nanodrop, 17.5 ng/µL --> worked
template
| restriction enzymes (biobrick assembly)
|
B0014 (from Christoph, verified PCR products, 21 ng/uL)
| EcoRI, PstI
|
R0011 (from PCR [15042010], 27.5 ng/µL
| SpeI
|
HisSig (1:100 dilution)
| XbaI
|
TrpSig (1:100 dilution)
| XbaI
|
| psB1K3 (with RFP insert, from HiWiPhilipp, 81 ng/µL)
| EcoRI, PstI
|
5 µL template used for each setup. protocol followed
- Gel for purification of the cleaved plasmid
- 2% Agarose in 1x TAE
- 120 V, 90 min
- stained with SybrGold
- digestion, digestion, 1 kb ladder
- Digestion worked (partly). band at 2000 bp (backbone) cut

- Purification of DNA from Gel
- Ligation of HisSig/TrpSig with R0011in 2 reactions
| used Volume
| approx. concentration*
|
HisSig
| 6 µL
| 7 ng/µL
|
TrpSig
| 6 µL
| 5 ng/µL
|
R0011
| 3 µL
| 6 ng/µL
|
* approximated from the amount used in the digestion before
Close
Week03
in vivo constructs
19.04.2010
- PCR of R0011-TrpSig and R0011-HisSig
- Purification with the Zymo Kit, Elution in 30 uL H2O
- Concentration measured with nanodrop: c(R0011-TrpSig)=20 ng/µL, c(R0011-HisSig)=12.5 ng/µl --> worked
- Gel for analysis of ligation and PCR
- 2% Agarose in 1x TAE
- 110 V, 90 min
- stained with SybrGold 1:10000 20 min
- pure R0011 PCR product used as control

LMW
| 4 µl
|
R0011-TrpSig
| 5 µL
|
R0011-HisSig
| 5 µL
|
R0011
| 5 µL
|
Samples seem to have run further than the buffer/dye-Front! But: Ligation Products show bands at shorter lengths than R0011 alone --> Ligation didn't work ?!?
- Ligation of HisSig/TrpSig with R0011in 2 reactions
| used Volume
| concentration
|
pSB1K3
| 5 µL
| 10 ng/µL (nanodrop)
|
B0014
| 3 µL
| 5 ng/µL approx.*
|
* approximated from the amount used in the digestion before
20.04.2010
- Gel for analysis of ligation and PCR (repeat of yesterday's gel)
- 2% Agarose in 1x TAE
- 130 V, 75 min
- stained with SybrGold 1:10000 60 min
- pure R0011 PCR product used as control
- Excision and purification of marked bands at 200 bp using QIA Kit, elution in 30 µl H2O

- PCR of excised and purified bands of R0011-TrpSig and R0011-HisSig
- complete samples (30 µl) used as templates
- Purification with the Zymo Kit, Elution in 30 uL H2O
- Concentrations of PCR-products: 0.5-1 ng/µl --> Gel excision or PCR didn't work
- Transformation (Woehlke-Lab)
- 8 µl of ligation product pSB1K3-B0014 to 50 µl XL-10 competent cells
- 200 µl plated on a Kanamycin-containing Plate
- remaining 800 µl stored @4°C in S1-lab
21.04.2010
- Gel for analysis of ligation and PCR (repeat of yesterday's gel)
- 2% Agarose in 1x TAE
- 110 V, 90 min
- stained with SybrGold 1:10000 80 min
- pure R0011 PCR product used as control
- Excision and purification of marked bands at 200 bp using Zymo 5 Kit, elution in 20 µl H2O
File:TUM2010 100421beschriftet.gif??
- PCR of excised and purified bands of R0011-TrpSig and R0011-HisSig
- complete samples (20 µl) used as templates
- Purification with the Zymo Kit, Elution in 25 uL H2O
- Concentrations of PCR-products:
- R0011-TrpSig: 22.5 ng/µl
- R0011-HisSig: 9.5 ng/µl
--> worked!!!!!
- Colony PCR
- 7 Colonies picked and resuspended in 20 µl LB+Kana (each)
- PCR of 2 µl of each sample, 2 µl as negative control (Program: ColonyPCR, modified)
- 15 µl of each sample mixed with 3 µl GLPn and loaded to Gel

- Overnight cultures:
- remaining 18 µl of samples 1, 3, 6, and 7 added to 5 ml LB + kanamycin
- 37°C on Shaker
22.04.2010
- Gel for purification of PCR products R0011-TrpSig and R0011-HisSig (yesterday's result)
- 2% Agarose in 1x TAE
- 110 V, 90 min
- stained with SybrGold 1:10000 30 min
- pure R0011 PCR product used as control
- Excision and purification of marked bands at 200bp using Zymo 5 Kit, elution in 20 µl H2O

- Miniprep
- Result: about 4 µg Plasmid
23.04.2010
template
| template volume
| restriction enzymes
| Buffer
|
HisSig (1:100 dilution)
| 5 µl
| EcoRI, SpeI
| NEB4
|
TrpSig (1:100 dilution)
| 5 µl
| EcoRI, SpeI
| NEB4
|
| psB1K3-B0014 from Miniprep (No 7, 35 ng/µl)
| 5 µl
| EcoRI, XbaI
| NEB4
|
Incubated 90 min @ 37°C
- Gel for purification of the cleaved plasmid
- 2% Agarose in 1x TAE (leftover from yesterday)
- 140 V, 90 min
- stained with SybrGold 40 min
- 4 µl 1 kb ladder, 10 µl purified digestion + 2 µl GLPn, 10 µl purified digestion + 2 µl GLPn
- Digestion worked (partly). band at 2400 bp cut out

- Purification of DNA from Gel
- A260/A230 and A260/A280 values were strange (see labbook)
Close
Week04
in vivo constructs
26.04.2010
- Digestion of pSB1K3-B0014 with EcorI and XbaI
- 10 µl template (No1, 50 ng/µl)
- 5 µl BSA, 5 µl Buffer NEB#4
- 1 µl EcoRI, 1 µl XbaI
- 28 µl H2O
- 1.5 h @ 37°C
- Purification with Zymo5 Kit, elution in 20 µl H2O
- Ligation of Signals and PSB1K3-B0014
- 3 µl of each sample, end volume 20 µl
- Preparation of Measurement Plasmid from Folder, Transformation
- Plate 1022, Spots 1E, 1G, 2A: pSB1A10 with different Inserts, all inserts are Zinc-finger constructs with about 1.6 kb
- Transformation of XL10 with Ligation Products (8 µl each) and pSB1A10 (2 µl each)
- Preparation of Measurement Plasmid from Folder, Transformation
- Plate 1022, Spots 1E, 1G, 2A: pSB1A10 with different Inserts, all inserts are Zinc-finger constructs with about 1.6 kb
- growing over night cultures of remaining PSB1K3-B0014-transformed cells
27.04.2010
- Plenty of cultures on both HisSig and TrpSig Ligation plates, but nothing on pSB1A10 plates! --> repeat DNA extraction, ask Prof. Simmel for new Distribution
- Colony PCR
- 7 Colonies picked and resuspended in 20 µl LB+Kana (each)
- PCR of 2 µl of each sample, 2 µl as negative control (Program: ColonyPCR, modified)
- 10 µl of each sample mixed with 2 µl GLPn and loaded to Gel
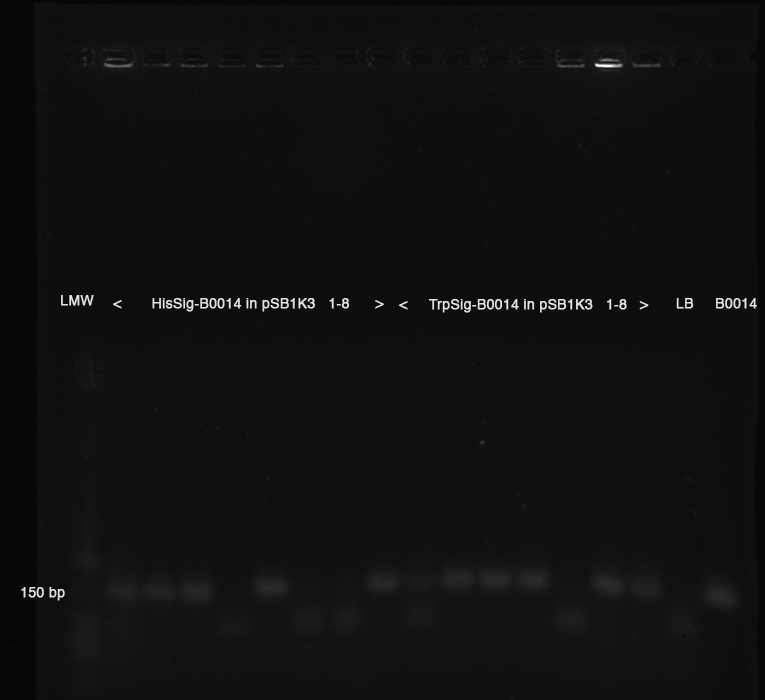
Many colonies with pSB1K3-B0014, not one with pSB1K3-Sig-B0014
- Miniprep of PSB1K3-B0014
- Samples I and II: 5 ml overnight cultures, centrifuged 10 min @ 3200 g, resuspended in 600 µl of the same culture
- Samples III and IV: 600 µl overnight cultures
- all samples mixed with 100 µl lysis buffer, Miniprep with Zyppy kit, each sample eluted in 50 µl H2O
- Concentration measured (Nanodrop, LP=1mm, Factor 10, 4 µl sample)
- cI=61.5 ng/µl
- cII=33.5 ng/µl
- cIII=103 ng/µl
- cIV=108 ng/µl
--> Better results for 600 µl cultures without centrifuging!!!
- Preparation of Measurement Plasmid from Folder, Transformation
- Plate 1022, Spots 1E, 1F, 1G, 1H, 2A: pSB1A10 with different Inserts, all inserts are Zinc-finger constructs with about 1.6 kb
28.04.2010
- No colonies on plates from Yesterday's transformations, but on the older plates (from monday) some colonies appeared
- 7 Colonies picked and resuspended in 20 µl LB0 (each)
- 1&2 from plate "1E", 3&4 from plate "1G", 5,6&7 from plate "2A", 8 LB0
- Colony PCR
- PCR of 2 µl of each sample, 2 µl as negative control (Program: ColonyPCR, modified)
- 15 µl of each sample mixed with 3 µl GLPn and loaded to Gel
- 1% Agarose in 1xTAE, 95 V, after 50 minutes changed to 110 V

- Digestion of pSB1K3-B0014 with EcorI and XbaI
- 10 µl template (No1, 50 ng/µl)
- 5 µl BSA, 5 µl Buffer NEB#4
- 1 µl EcoRI, 1 µl XbaI
- 28 µl H2O
- 1.5 h @ 37°C
- heat inactivation 5min @60°C
- Dephosphorylation of restricted vector
- Purification with Zymo5 Kit, elution in 20 µl H2O
- loaded on gel (with 4 µl GLPn) (Gel shown above)
- Gel excision with Zymo Kit
29.04.2010
- Digestion of pSB1K3-B0014 with EcorI and XbaI
- 10 µl template (NoIV, 108 ng/µl)
- 5 µl BSA, 5 µl Buffer NEB#4
- 1 µl EcoRI, 1 µl XbaI
- 28 µl H2O
- 1.5 h @ 37°C
- heat inactivation 5min @60°C
- Dephosphorylation of restricted vector
- Purification with Zymo5 Kit, elution in 20 µl H2O
- loaded on gel (with 4 µl GLPn)
File:X
- Gel excision with Zymo Kit
- Digestion of R0011 with SpeI
- 10 µl template (R0011, X ng/µl)
- 5 µl BSA, 5 µl Buffer NEB#4
- 1 µl SpeI
- 29 µl H2O
- 1.5 h @ 37°C
- Ligation
- 5 µl R0011 (S-digested) with 12 µl TrpSig or HisSig, respectively (X-digested)
- complete ligation (20 µl) loaded on Gel (with 4 µl GLPn)
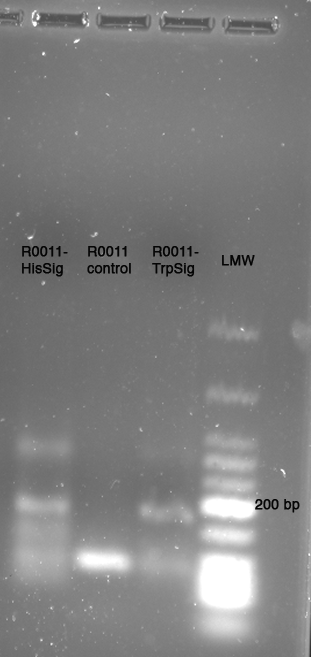
- Gel excision with Zymo Kit, eluted in 42 µl H2O
- Transformation
- 50 µl XL-10 transformed with 2 µl of pSB1A10 prepared from IGem 2009 Distribution (13 µl left in pink Box @-20°C)
-
30.04.2010
PCR R0011-HisSig and R0011-TrpSig --> 13 ng/µl x 20 µl
Close
Week05
in vivo constructs
04.05.2010
- Digestion of pSB1K3-B0014 with EcorI and XbaI
- 10 µl template (NoIII, 103 ng/µl)
- 5 µl BSA, 5 µl Buffer NEB#4
- 1 µl EcoRI, 1 µl XbaI
- 28 µl H2O
- 1.5 h @ 37°C
- Purification with Zymo5 Kit, elution in 15 µl H2O
- loaded on gel (with 3 µl GLPn)

- Gel excision with Zymo Kit
- Digestion of R0011-HisSig and R0011-TrpSig with EcorI and SpeI
- 10 µl template (PCR-product)
- 5 µl BSA, 5 µl Buffer NEB#4
- 1 µl EcoRI, 1 µl SpeI
- 28 µl H2O
- 1.5 h @ 37°C
- Purification with Zymo5 Kit, elution in 20 µl H2O
- Ligation
- 4 µl R0011-Signal (E/S-digested) with 4 µl pSB1K3-B0014 (E/X-digested)
- 15 min @ RT, 20 min heat inactivation @ 65°C
- Overrnight liquid cultures of pSB1A10-RFP made from
--> each in 600 µl LB+Carbenicillin (=Ampicillin) @37°C
05.05.2010
- Miniprep of pSB1A10; 4 samples (1, 2; 3a; 3b)
- eluted in 50 µl H2O each
- Concentrations:
- c1=37.5 ng/µl
- c2=56.5 ng/µl
- c3a=46.5 ng/µl
- c3b=30 ng/µl
- Digestion of pSB1A10 with EcorI and PstI, 4 samples (1, 2; 3a; 3b)
- 15 µl template
- 5 µl BSA, 5 µl Buffer NEB#3
- 1 µl EcoRI, 1 µl PstI
- 23 µl H2O
- 1.5 h @ 37°C
- heat inactivation 5min @60°C
- Purification with Zymo5 Kit, elution in 15 µl H2O
- loaded on gel (with 3 µl GLPn)
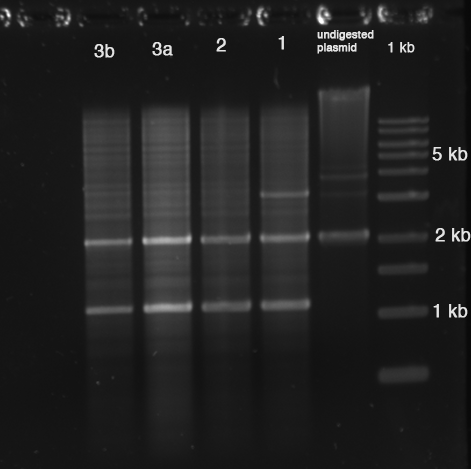
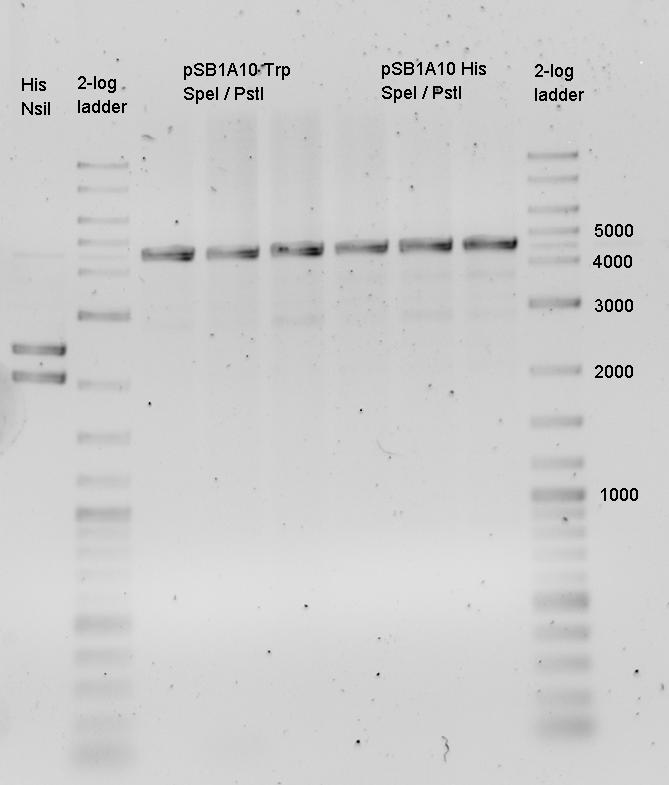
Insert @ 1 kb as expected, but vector @ 2 kb and not @ 5 kb as expected!!!!
--> Wrong Plasmid! Comparison to the [http://partsregistry.org/cgi/assembly/plate_egel.cgi?id=615 Gel in the registry] shows: The Distribution contains the wrong plasmid!
- Digestion of HisTerm and TrpTerm with EcorI and PstI
- 5 µl template
- 5 µl BSA, 5 µl Buffer NEB#3
- 1 µl EcoRI, 1 µl PstI
- 33 µl H2O
- 1.5 h @ 37°C
- Clones picked: 7 from each Plate (pSB1K3-R0011-TrpSig-Boo14 and pSB1K3-R0011-HisSig-Boo14)
- Colony PCR
- PCR of 2 µl of each sample, 2 µl as negative control (Program: ColonyPCR)
- 15 µl of each sample mixed with 3 µl GLPn and loaded to Gel
- 2% Agarose in 1xTAE, 130 V, 90 min

06.05.2010
- Digestion of pSB1K3 with EcorI and XbaI
- 20 µl template (sample III, 103 ng/µl)
- 5 µl BSA, 5 µl Buffer NEB#3
- 1 µl EcoRI, 1 µl XbaI
- 18 µl H2O
- 1.5 h @ 37°C
- heat inactivation 5min @60°C
- loaded on gel (with 10 µl GLPn) in 4 lanes

- Gel excision with Zymo Kit (lanes 1&2) and with Qiaquick Kit (lanes 3&4)
- c1=4.5 ng/µl
- c2=3.5 ng/µl
- c3=2 ng/µl
- c1=7 ng/µl
- A260/A230 and A260/A280 values were strange (see labbook)
- Ligation
- 4 µl R0011-Signal (E/S-digested) with 10 µl pSB1K3-B0014 (E/X-digested, from 23.04.)
- 15 min @ RT, 20 min heat inactivation @ 65°C
07.05.2010
- Clones picked: 7 from each Plate (pSB1K3-R0011-TrpSig-Boo14 and pSB1K3-R0011-HisSig-Boo14)
---Too damn stupid to do a PCR!!!---
- replated picked clones on new plates, incubated at RT
Close
Week06
in vivo constructs
10.05.2010
- Colony PCR of picked clones from Fr 07.05.2010
- PCR of 2 µl of each sample, 2 µl as negative control (Program: ColonyPCR, modified)
- 15 µl of each sample mixed with 3 µl GLPn and loaded to Gel
- 2% Agarose in 1xTAE, 120 V, 110 min
- stained in SybrSafe 50 min

Interpretation:
Colonies contain an Insert with Prefix and Suffix, length is 200 bp. This is too short for the desired R0011-Signal-B0014 (245 or 247 bp) construct, but longer than B0014 (136 bp) which was the Insert in the digested vector.
Possible explanation: Ligation worked, but not with R0011-Signal-construct but with R0011 or Signal. Which?
| fragment
| length without Prefix/Suffix
| length with Prefix/Suffix
|
| R0011
| 55 bp
| 96 bp
|
| B0014
| 95 bp
| 136 bp
|
| TrpSig/HisSig
| 34 bp/32 bp
| 75 bp/73 bp
|
| TrpSig-B0014/HisSig-B0014
| 135 bp/ 133 bp
| 176 bp/ 174 bp
|
| R0011-TrpSig-B0014/R0011-HisSig-B0014
| 206 bp/ 204 bp
| 247 bp/ 245 bp
|
| R0011-B0014
| 156 bp
| 197 bp
|
| R0011-TrpSig/R0011-HisSig
| 95 bp/93 bp
| 136 bp/134 bp
|
Prefix: 20 bp; Suffix: 21 bp; X-S-scar: 6 bp
--> it looks as if R0011 is ligated to B0014, which makes the whole construct wothless. The R0011-HisSig control looks more like R0011 alone as well.
11.05.2010
- Ligation
- 4 µl Signal (E/S-digested; from ) with 5 µl pSB1K3-B0014 (E/X-digested; from)
- 15 min @ RT
12.05.2010
- Colony PCR of picked clones from Tu 12.05.2010
- PCR of 2 µl of each sample, 2 µl as negative control (Program: ColonyPCR, modified)
- 15 µl of each sample mixed with 3 µl GLPn and loaded to Gel
- 2% Agarose in 1xTAE, 120 V, 110 min
- stained in SybrSafe 60 min
File:TUM2010 100512beschriftet.png
| fragment
| length without Prefix/Suffix
| length with Prefix/Suffix
| length after PCR
|
| R0011
| 55 bp
| 96 bp
| 104 bp
|
| B0014
| 95 bp
| 136 bp
| 154 bp
|
| TrpSig/HisSig
| 34 bp/32 bp
| 75 bp/73 bp
| 93 bp/91 bp
|
| TrpSig-B0014/HisSig-B0014
| 135 bp/ 133 bp
| 176 bp/ 174 bp
| 194 bp/ 192 bp
|
R0011-TrpSig-B0014/
R0011-HisSig-B0014
| 206 bp/ 204 bp
| 247 bp/ 245 bp
| 265 bp/ 263 bp
|
| R0011-B0014
| 156 bp
| 197 bp
| 215 bp
|
| R0011-TrpSig/R0011-HisSig
| 95 bp/93 bp
| 136 bp/134 bp
| 154 bp/152 bp
|
Prefix: 20 bp/29 bp after PCR; Suffix: 21 bp/30 bp after PCR; X-S-scar: 6 bp
14.05.2010
- Digestion of pSB1K3 with EcorI and XbaI
- 10 µl template (sample III, 103 ng/µl)
- 2 µl BSA, 2 µl Buffer NEB#4
- 1 µl EcoRI, 1 µl XbaI
- 4 µl H2O
- 1 h @ 37°C
- loaded on gel (with 4 µl GLPn) in 1 lane
File:TUM2010 100514beschriftet.png
- Gel excision with Zymo Kit
- Digestion of HisSig and TrpSig with EcorI and SpeI
- 10 µl template ("1:100")
- 2 µl BSA, 2 µl Buffer NEB#3
- 1 µl EcoRI, 1 µl SpeI
- 4 µl H2O
- 1.5 h @ 37°C
- Purification with Zymo 5
- or heat inactivated (20 min @ 80°C)
- Transformation
- 50 µl XL-10 transformed with 10 µl of Ligation mix
- 50 µl untransformed cells plated on Kana-plate as control
Close
Week07
in vivo constructs
17.05.2010
- Plates from Friday:
- plenty colonies on control plate --> XL10 cells are impure!
- use DH5a from now on!!!!!
- Transformation
- 50 µl DH5a transformed with 10 µl of Friday's Ligation mix
- plated on Kana-Plates; Overnight @ 37°C
- DNA Isolation from BioBrick Distribution 2010
- 10 µl H2O added to Well 1A of plate 1 containing pSB1A10 with RFP-insert
- 2 µl used for Transformation of 50 µl DH5a-cells
- plated on Carbenicillin (=Amp-analogon)-plates, Overnight @ 37°C
18.05.2010
- Colony PCR of picked clones
- PCR of 2 µl of each sample, 2 µl as negative control (Program: ColonyPCR)
- 10 µl of each sample mixed with 10 µl Formamide loading buffer and loaded to Polyacrylamide Gel
Samples:
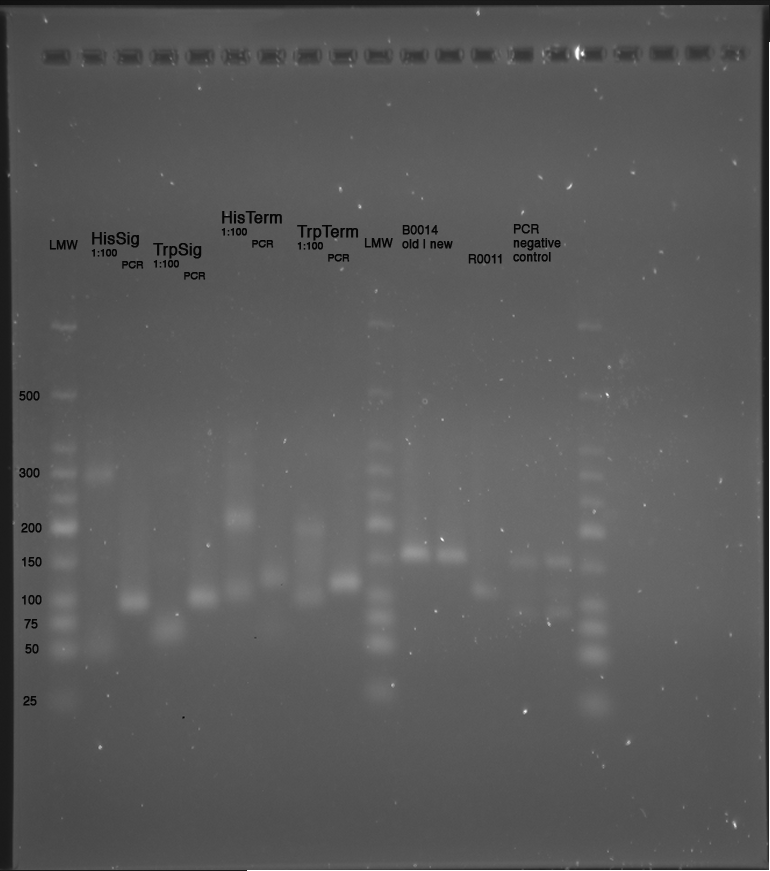
LMW|R0011|HisSig|TrpSig|HisSig E/S-Dig|TrpSig E/S-Dig|B0014|LMW2|Colony PCR His1|His2|Trp1|Trp2|control|HisTerm|TrpTerm|HisTerm E/P-Dig|TrpTerm E/P-Dig
- 5 µl of each samples mixed with 5 µl formamide loading dye and loaded to gel(except Ladder and colonyPCR)
- LMW: 3 µl LMW (Korbinian) + 3 µl Formamide loading Dye
- LMW2: 5 µl LMW Quickload (with GLP) + 10 µl Formamide loading Dye
- stained in SybrSafe
Important Mistake! See below Gel!

IMPORTANT MISTAKE: DENATURING GELS NOT USEFUL FOR dsDNA!!!
REPEAT WITH NATIVE GEL, IGNORE INTERPRETATION!!!
(*R0011 and B0014 look normal
- ColonyPCR: bands that look like B0014 in all clones (and in control???? strange!) --> Religation?
- Signals at the wrong size: should be about 75 bp, look like 200 bp!!!
- terminators completely strange: should be around 100 bp!
--> are all of our sequences just wrong???
What are we going to do? Order everything new?)
19.05.2010
- Gel from Korbinian
- 5 µl of each samples mixed with 5 µl formamide loading dye and loaded to gel(except Ladder and colonyPCR)
- LMW: 3 µl LMW (Korbinian) + 3 µl Formamide loading Dye
- colony PCR: from Tuesday, 8 µl sample with 8 µl Formamide loading Dye
- stained in SybrSafe 20 min

- Colony PCR
- 4 colonies picked from each Plate (Ligations from yesterday; Signal-B0014)
- 15 µl of each Sample mixed with 3 µl GLPn and loaded to Gel:
- 3% Agarose in 1x TBE, 130 V
File:TUM2010 100519beschriftet.png600px
20.05.2010
- Digestion
- template
- 2 µl BSA
- 2 µl Buffer
- 1 µl of each enzyme
- water to reach 20 µl
Template
| Enzymes
| NEB Buffer #
|
HisTerm & TrpTerm (10 µl)
| EcoRI, PstI
| 3
|
HisSig & TrpSig (10 µl)
| EcoRI, SpeI
| 4
|
B0014 (5 µl)
| XbaI, PstI
| 3
|
pSB1A10_RFP (14 µl)
| EcoRI, PstI
| 3
|
pSB1K3_RFP (14 µl)
| EcoRI, PstI
| 3
|
pSB1K3_B0014 N° 4 (14 µl)
| EcoRI, XbaI
| 4
|
- incubated @37°C for 1.5 h
- digested inserts heat inactivated (20 min @ 80°C)
- digested plasmids loaded on gel (with 4 µl GLPn) in 1 lane
- Gel excision with Zymo Kit
- c(pSB1A10, I)=4.5 ng/µl
- c(pSB1A10, II)=1.5 ng/µl ?!?!?!
- c(pSB1K3 E/P)=7 ng/µl
- c(pSB1K3_B0014 E/X)=2.5 ng/µl
- Gel of PCR products
- 3% Agarose in 1x TBE; 2h @130 V
- Ligation
- templates
- 2 µl T4-buffer 10x
- 1 µl T4-Ligase
- Water to reach 20 µl
Vector
| Insert
|
psB1A10 (E/P; sample I) (10 µl)
| TrpTerm (E/P) (4 µl)
|
psB1A10 (E/P; sample I) (10 µl)
| HisTerm (E/P) (4 µl)
|
psB1K3_B0014 (E/X) (12 µl)
| HisSig (E/S) (2 µl)
|
psB1K3_B0014 (E/X) (12 µl)
| TrpSig (E/S) (2 µl)
|
psB1K3 (E/P) (8 µl)
| HisSig (E/S)(2 µl) + B0014 (X/P)(1.5 µl)
|
psB1K3 (E/P) (8 µl)
| TrpSig (E/S)(2 µl) + B0014 (X/P)(1.5 µl)
|
- Transformation
- 50 µl DH5a transformed with 10 µl of Ligation mix
- 50 µl DH5a transformed with 2 µl of pSB1K3_B0014
- 50 µl DH5a transformed with 2 µl of pSB1K3_RFP
- 50 µl DH5a transformed with 2 µl of pSB1A10_RFP
21.05.2010
- Colony PCR
- 4 colonies picked from each Plate (pSB1K3_HisSig_B0014, pSB1K3_TrpSig_B0014, pSB1K3_HisSig_B0014 double ligation, pSB1K3_TrpSig_B0014 double ligation)
- each clone resuspended in 20 µl LB0, 3 µl used as template for PCR
- 15 µl of each Sample mixed with 3 µl GLPn and loaded to Gel:
- 3% Agarose in 1x TBE, 130 V

Close
Week08
in vivo constructs
25.05.2010
- Colony PCR
- 4 colonies picked from each Plate
- pSB1K3_HisSig_B0014
- pSB1K3_TrpSig_B0014
- pSB1K3_HisSig_B0014 double ligation
- pSB1K3_TrpSig_B0014 double ligation
- pSB1A10_TrpTerm
- pSB1A10_HisTerm
- each clone resuspended in 20 µl LB0, 2 µl used as template for PCR
- 15 µl of each Sample mixed with 3 µl GLPn and loaded to Gel:
- 3% Agarose in 1x TBE, 220 V (double Gel, 35 cm)
- stained in SybrSafe
600px

- overnight cultures made of
- HisSig 3, DL1, DL4
- TrpSig DL2, DL4
- HisTerm/TrpTerm 1,2,3
26.05.2010
- Miniprep of cultures set up 25.05.2010
- HisSig 3, DL1, DL4
- TrpSig DL2, DL4
- HisTerm/TrpTerm 1,2,3
- Restriction
- analytical: E/P HisSig(3, DL2); TrpSig(DL2, DL4): 1.5 h 37 °C
- prep: E/X HisSig(3, DL2); TrpSig(DL2, DL4): 1.5 h 37 °C
- prep: E/S R0011: 1.5 h 37 °C, inactivation 20 min @ 80 °C
- total volume each 20 µl, 10 µL template
- Gel: 1% Agarose, TAE - 1,5 h 110 V
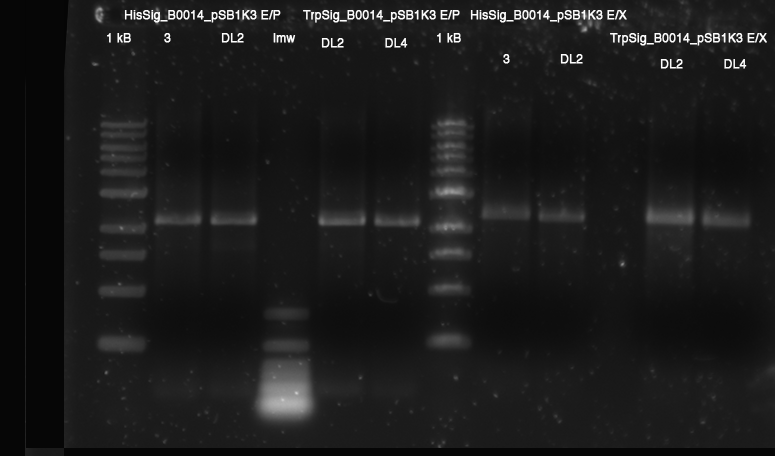
- Ligation:
- HisSig_3 (E/X) with R0011 (E/S)
- HisSig_DL2 (E/X) with R0011 (E/S)
- TrpSig_DL2 (E/X) with R0011 (E/S)
- TrpSig_DL4 (E/X) with R0011 (E/S)
- batches
- total volume 20 µL
- 2 µL R0011 (E/S)
- 2 µL T4 buffer
- 1 µL T4 Ligase
- 15 µL vector
- PCR
- His and TrpSig
- His and TrpTerm
- R0011
- B0014
- 50 µL total volume
- 1 µL template
- 1 µl G1004
- 1 µl G1005
- 0.2 µL Taq
- 5 µl Taq standard buffer
- rest water
27.05.2010
- Colony PCR
- 3 colonies picked from each Plate
- pSB1K3_R0011_HisSig_B0014 (N° 3 from yesterday)
- pSB1K3_R0011_HisSig_B0014 (N° DL1 from yesterday)
- pSB1K3_R0011_TrpSig_B0014 (N° DL2 from yesterday)
- pSB1K3_R0011_TrpSig_B0014 (N° DL4 from yesterday)
- "N6"
- "N15"
- each clone resuspended in 20 µl LB0, 2 µl used as template for PCR
- 15 µl of each Sample mixed with 3 µl GLPn and loaded to Gel:
- 3% Agarose in 1x TBE, 220 V (double Gel, 35 cm)
- stained in SybrSafe
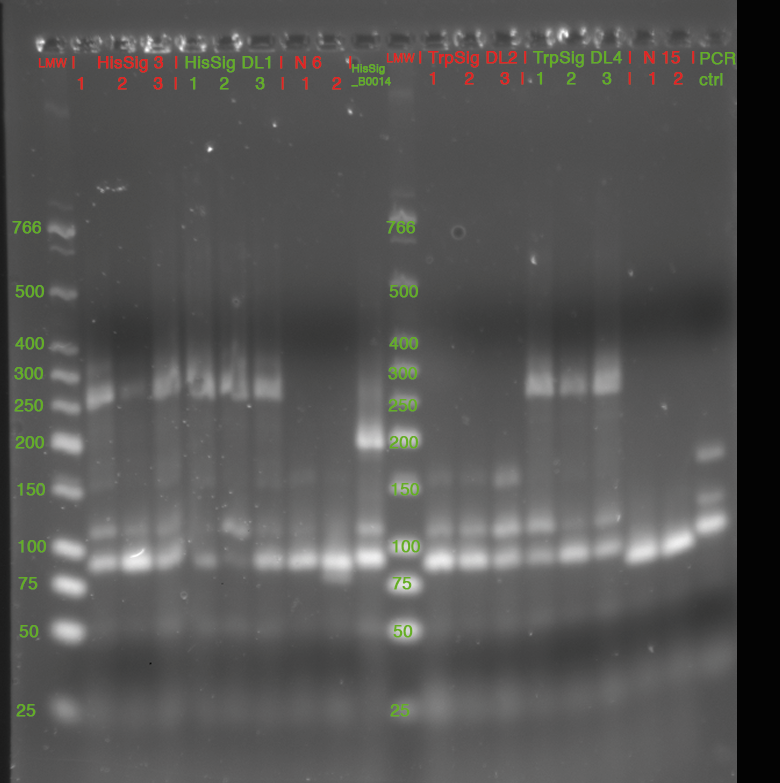
| fragment
| length without Prefix/Suffix
| length after PCR
|
| R0011
| 55 bp
| 104 bp
|
| B0014
| 95 bp
| 154 bp
|
| TrpSig/HisSig
| 34 bp/32 bp
| 93 bp/91 bp
|
| TrpSig-B0014/HisSig-B0014
| 135 bp/ 133 bp
| 194 bp/ 192 bp
|
R0011-TrpSig-B0014/
R0011-HisSig-B0014
| 206 bp/ 204 bp
| 265 bp/ 263 bp
|
|
|
|
|
| I712074 ("N6")
| 46 bp
| 105 bp
|
| I719005 ("N15")
| 23 bp
| 82 bp
|
Prefix: 29 bp after PCR; Suffix: 30 bp after PCR; X-S-scar: 6 bp
- overnight cultures made of
- HisSig 3_1, DL1_3
- TrpSig DL4_1, DL4_3
- N15 1&2
- HisTerm/TrpTerm (picked colonies from yesterdays plates #2 each)
- Purification of yesterday's PCR
- elution in 50 µl H2O
- c(HisSig)=5.5 ng/µl
- c(TrpSig)=10 ng/µl
- c(HisTerm)=6.5 ng/µl
- c(TrpTerm)=6.5 ng/µl
- c(R0011)=13 ng/µl
- c(B0014)=12.5 ng/µl
- 5 µl loaded on gel with 1 µl GLPn; 3% Agarose in 1x TBE, 220 V (double Gel, 35 cm)
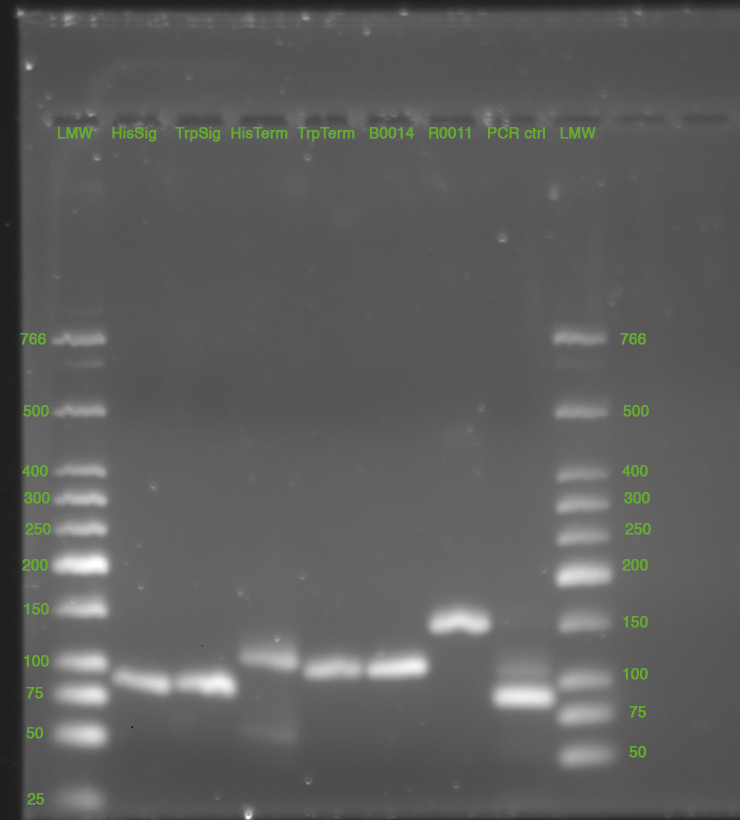
28.05.2010
- Miniprep of cultures from 27.05.2010, Elution in 50 uL nuclease free water
- (1)HisSig 3_1
- (2)HisSig DL1_3
- (3)TrpSig DL4_1
- (4)TrpSig DL4_3
- (7)HisTerm#1
- (8)HisTerm#2 (7 ml culture)
- (9)TrpTerm#1
- (10)TrpTerm#2 (7ml culture)
- (5)N15-1 (=BBa_I719005)
- (6)N15-2 (=BBa_I719005)
sample
| DNA concentration (ng/uL)
|
HisSig 3_1
| 6
|
HisSig DL1_3
| 11
|
TrpSig DL4_1
| 16
|
TrpSig DL4_3
| 21.5
|
HisTerm#1
| 27
|
HisTerm#2
| 66
|
TrpTerm#1
| 34
|
TrpTerm#2
| 29.5
|
N15-1 (=BBa_I719005)
| 19.5
|
N15-2 (=BBa_I719005)
| 18.5
|
- analytical digest (E/P)
- of all samples. total volume 20 uL, 5 uL template used for Term-constructs, 10 uL teplate for all others
- Agarose Gel
- 3% broad range agarose in TBE. Run in TBE, 140 V, 1.50 h
- stained with SybrGold, 45 min
- signals look fine
- terminators also (without pre/suffix 97/104 bp)
- T7 promoter without pre/suffix has a length of 23 bp + cut pre/szffix ca at 60 bp -->buffer?

Close
Week09
in vivo constructs
31.05.2010
- Digestions
- 15N-1 (BBa_I719005, 19.5 ng/uL) with S/P
- HisSig/TrpSig (6.5/10 ng/uL) with X/P
- 2 h 37 °C
- heat inactivation of insert-digestions
- Gel: 1% Agarose in 1x TAE
- 1 h 25 min, 115 V
- stained with SybrGold, 40 min

- Band at ~2100b cut und purified using the zymo kit
- ligation
- HisSig (4 µL of digest) with purified plasmid (with BBa_I719005)
- TrpSig (2 µL of digest) with purified plasmid -=-
- reason: concentration of His Sig before digest was 1/2 of TrpSig
- of DH5a with Ligation batches, HisSig1-3, HisSig3-1, TrpSig DL4-1, TrpSig 4-3
01.06.2010
- of Ligations transformed into DH5a yesterday
- 4 Colonies of each Ligation
- Gel: 3% broad range Agarose in 1xTBE
- 1.5 h 140 V
- stained with SybrGold

- calculation for the expected size of the fragments
| part
| size (bp)
|
| HisSig
| 32
|
| TrpSig
| 34
|
| T7 promoter
| 23
|
| prefix
| 20
|
| suffix
| 21
|
| X/S scar
| 6
|
- in PCR we get additional bp due to the primers - +9 at pre/suffix=+18 bp
- overall size of the fragments expected to come out of the PCR: T7_HisSig: 120 bp, T7_TrpSig: 122 bp
- 5 ml cultures of pSB1K3_R0011_HisSig_B0014 (1_3 & 3_1) and pSB1K3_R0011_TrpSig_B0014 (DL4_1 & DL4_3)
- 1 ml cultures of each colony monitored in Colony PCR
02.06.2010
- Miniprep of yesterdays cultures using Zymokit, elution by nuclease-free water
- Concentration determination
- analytic digestion
- results on gel:
JobNr. Barcode Last change Date/Time Last message / Files 6549287 AE2739 02.06.2010 / 13:51:12 HisSig 1-3-forward G1004
We just received your order. Many thanks.
6549288 AE2738 02.06.2010 / 13:51:12 HisSig 3-1-forward G1004
We just received your order. Many thanks.
6549289 AE2737 02.06.2010 / 13:51:12 TrpSig DL4-1-forward G1004
We just received your order. Many thanks.
6549290 AE2736 02.06.2010 / 13:51:12 TrpSig DL4-3-forward G1004
We just received your order. Many thanks.
6549291 AE2735 02.06.2010 / 13:51:12 HisTerm-forward G1004
We just received your order. Many thanks.
6549292 AE2734 02.06.2010 / 13:51:12 TrpTerm-forward G1004
We just received your order. Many thanks.
- Gel 3% broad range agarose in 1x TBE
-

Close
Week10
in vivo constructs
07.06.2010
- Sequenbcing results from GATC
- HisSig DL1-3 is ok
- HisSig 3-1 is ok
- TrpSig DL4-1 is ok
- TrpSig DL4-3 is ok
- TrpTerm + HisTerm bad runs... --> new sequencing order with Primer 100 bp upstream (within GFP)
- Files can be found stored in our GATC account
- Sequencing@GATC: both Term-constructs with primer pGFP-FP provided by GATC
- Restrictions
- psB1A10_TrpTerm/HisTerm with Nsi1, Aat2
- pSB1K3_R0011_HisSig/TrpSig_B0014 with Pst1, Aat2
- T7 bb with Spe1, Pst1
- PCRProducts: HisSig/TrpSig with Pst1, Xba1, 2 h @37°C
- all plasmid digests done sequential as enzymes do not have 100% activity in the same buffer, each reaction 1.5 h@37°C
- psB1A10_TrpTerm/HisTerm and T7 bb dephosphorylated the last 30 min
liquid culture (10 ml) of pSB1K3_R0011_HisSig/TrpSig_B0014
08.06.2010
- Sequencing results from GATC: His/TrpTerm with pGFP-FP primer
- HisTerm worked
- TrpTerm worked
- checked with blast2seq
- 8 colonies from each plate of T7His, T7Trp, MonsterHis, MonsterTrp as ligations resulted in many colonies
- for all colony PCR reactions


Interpretation/Info: The R0011_Sig_B0014 construct was cut with PstI, but not ligated into a PstI site but instead into the NsiI site --> even if sticky end are compatible, the bases in the 3` direction are different --> primer lags 7 bp compared to standard procedure --> we didn´t expect to find the signal construct by colony PCR --> control digestion tomorrow
- T7-Signal constructs seem to have worked, expected size was 23 bp (T7)+ 32 (HisSig)/34 (TrpSig) bp + 30+29 (PCRPre+Suf)=114/116 bp
09.06.2010
- Miniprep of 4 Monster_His, 4 Monster_Trp, 3 T7_His and 3 T7_Trp cultures
- Analytical digestion of plasmids mentioned above
- gel of Monster_Plasmid digestion

gel didn´t work at all --> even after > 2 h, bands were not separated correctly, even the 1kb ladder was "stacked" in the gel-pockets, the 100 bp ladder should show EQUAL distances between the lines [http://www.neb.com/nebecomm/productfiles/778/images/N3231_fig1_v1_000034.gif see here], it looks like the gel was "more dense" at the pockets---> no idea what happened --> repeat Monster-digestion tomorrow?
- gel of T7_Plasmid digestion
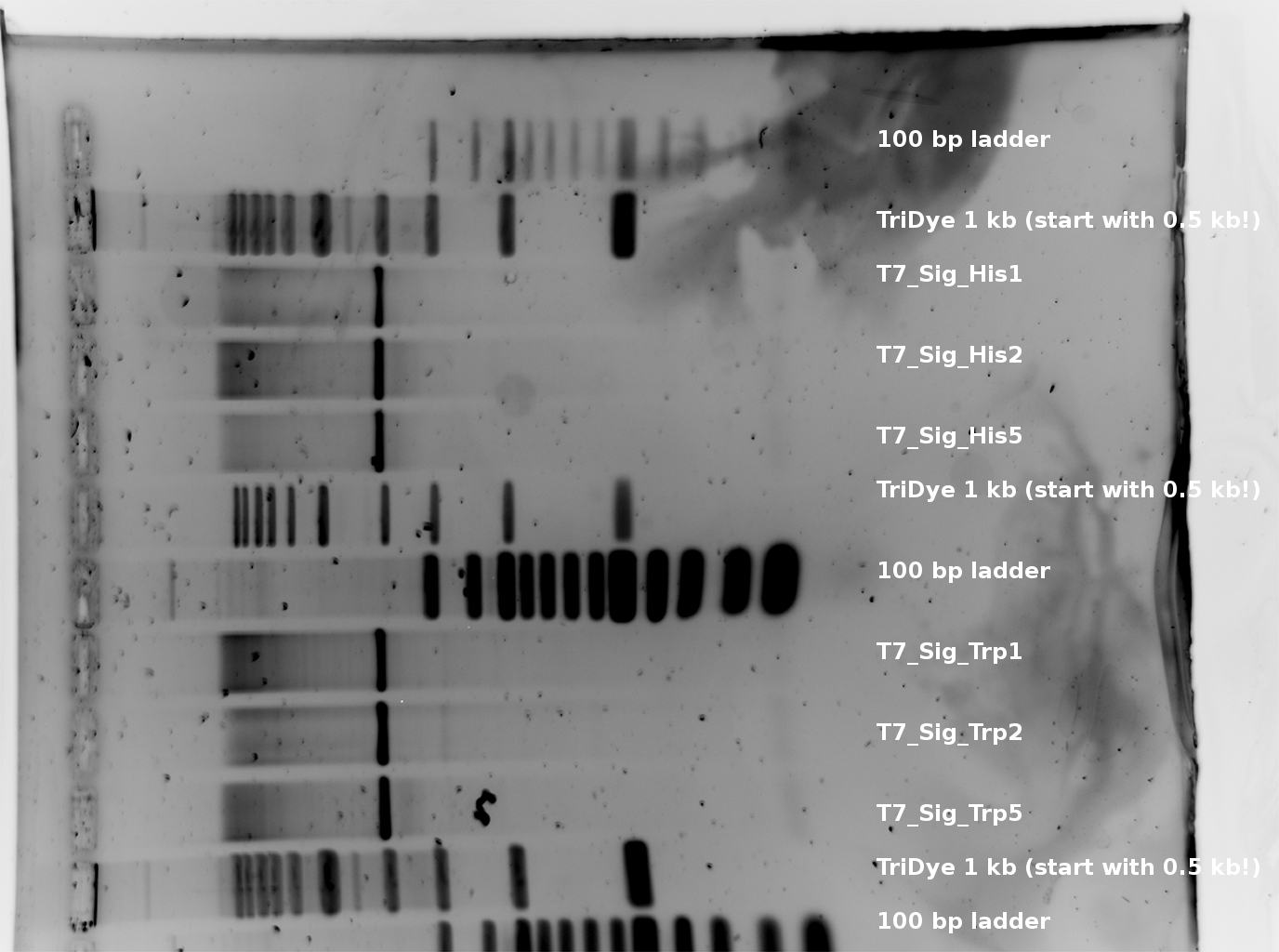
T7_Trp E + S digestion 107 bp and T7_His 105 bp --> worked for all picked colonies. (regard that there is an excess of plasmid DNA-basepairs of factor >30 --> thats why the inserts are much weaker than the plasmid signals.
occured trouble:
- Ladders and loading dye´s empty --> i used those of eike, BUT: eikes 1 kb ladder is different --> compare [http://www.neb.com/nebecomm/products/productN3272.asp here] and his loading dye was much more diluted, even if there was also 6x Sac GLP written on it -> i hope this won´t cause any trouble
10.06.2010
- Promega E.coli S30 in vitro transcription/translation kit
- Spe1, Aat2 from NEB, 500 U each
- of Ligation colonies from MonsterHis/trp 1-3 and T7His/Trp 1,2,5/1,2,3
- 2h digestion
- Monster: 6 µL DNA template with Aat2/Spe1 in Buffer 4/Bsa
- T7-Signal: 6 µL DNA template with E/P in Buffer 3
- used standards: lmw, 2-log click here
- Gel1: 1% Agarose in 1xTBE for Digestions of Monsterplasmid
- run in big chamber @ 200 V for 1 h 20 min

- Gel2: 3% Agarose (broad range) in 1xTBE for Digestions of T7-Signal
- run in small chamber @140 V for 1 h 35 min

- Conclusions:
- all T7-Signal ligations loaded on the gel worked
- monsterplasmid didn't work? bands at 800 bp, 900 bp, 1.3 kbp, 2.2 kbp, 3 kbp, we SHOULD expect to see our Insert, wich is Prefix+R0011_Signal_B0014_small Suffix, which should run around 300-400 bp...
11.06.2010
- Gel: large 1% Agarose in TAE. Load: The rest of N/A cut Messplasmids from 07.06.2010. Run @220 V for 3.5 h
- fragments expected are 5087 and 176. original size of plasmid is 5263. This is a Try to differ between 5087 and 5263 bp

- Band @ 5000 bp of Trp_Term purified, obviously digestion was 100%. Bad point is that HisTerm includes an Nsi1 cleavage site...
Close
Week11
in vivo constructs Promega Kit
14.06.2010
- 10 µL pSB1A10_TrpTerm Aat2/Nsi1 0.5 ng/uL
- 1 µL R0011_TrpSig_B0014 Aat2/Psb1 11 ng/uL
- 2 µL T4 ligase buffer
- 1 µL T4 Ligase
- 6 µL H2O
- 10 min RT
- Transformation of DH5a with
- Ligation
- T7His#1
- T7Trp#1
- pSB1K3_R0011_TrpSig_B0014
- pSB1K3_R0011_HisSig_B0014
- pSB1A10_TrpTerm
- pSB1A10_HisTerm
15.06.2010
- Over night cultures
- Aliquots of the Promega in vitro expressions kit from E. coli S30 extract:
- 40 µL with aa mix including all aa.
16.06.2010
Fluoresence measurements using in vitro kit
- in vitro kit sample
- adding psBA1A10 Trp_Term --> constant over time, no significant changes compared to kit alone --> high efficiency of AraC
- adding L-(+)Arabinose (final concentration 2%) --> after approx. 10 min significant GFP production --> measuring for xxx min --> RFP is slightly increased (to proof if correlated to GFP peak --> crossdetection)
- adding psB1K3 R0011_TrpSig_B0014
Cell culture
5 ml culture for
- psBA1A10 Trp_Term/HisTerm
- psB1K3 R0011_TrpSig/Hissig_B0014
17.06.2010
Cloning
Digestion of Trp-Sig with E/P and psB1A10 Trp_Term with E/P
- Gel purification of psB1A10 Trp_Term E/P cut
- Heat inactivation of Trp-Sig E/P cut
- Ligation for 10 min @ RT and Transformation in DH5-a cells
18.06.2010
cloning
- Transformation (about 20 colonies) --> picking 5 colonies
- colony PCR
- Gel
2 % broad range agarose, 1 h 120 V 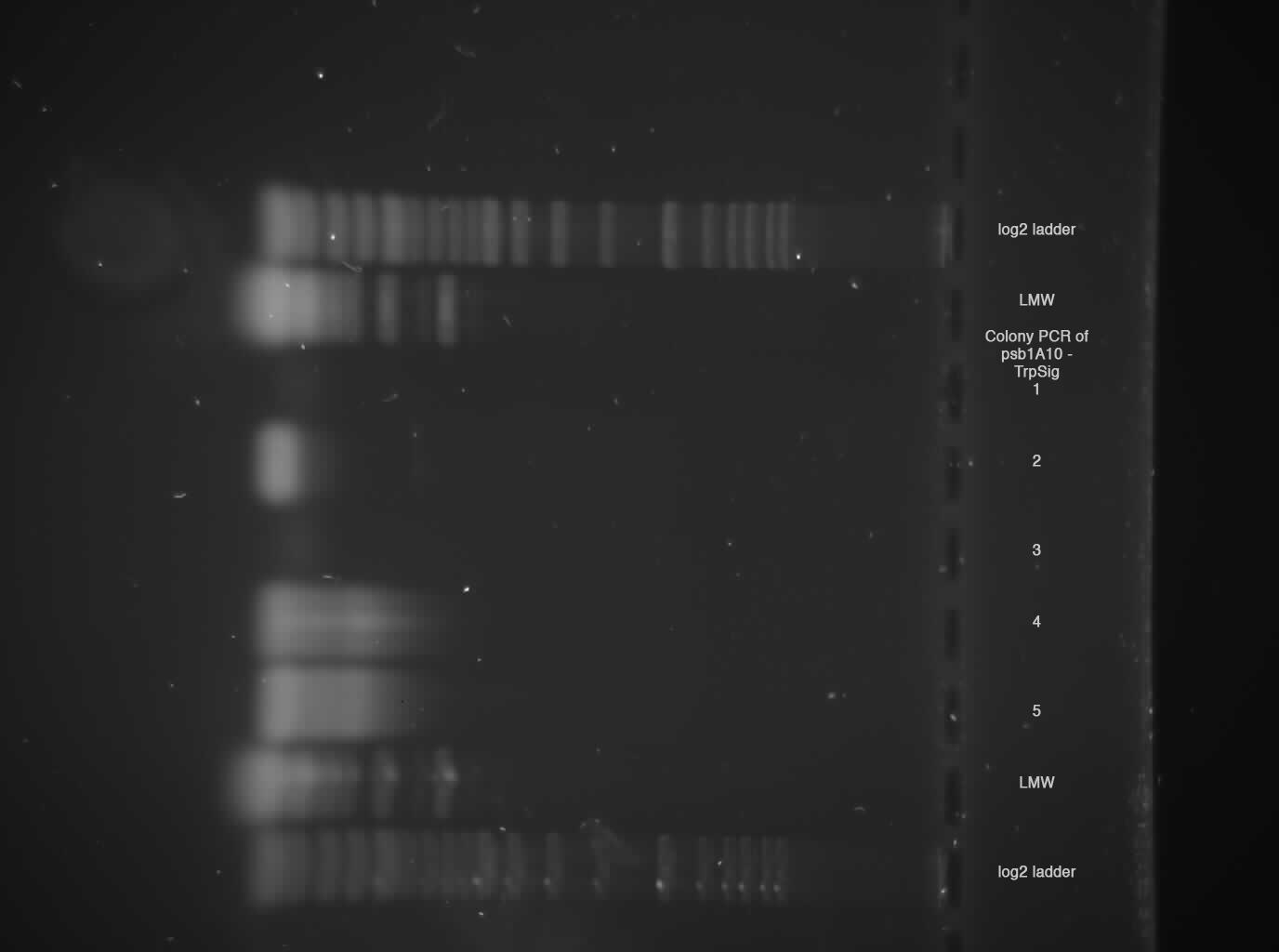 Sample 2, 4, 5 shows probably Trp-Signal + Pre/Suffix --> send sample 2 for sequencing!
Sample 2, 4, 5 shows probably Trp-Signal + Pre/Suffix --> send sample 2 for sequencing!
- control digestion of all 10 picked psB1A10-TrpSig in 1% broad range agarose, > 3 h, 120 V
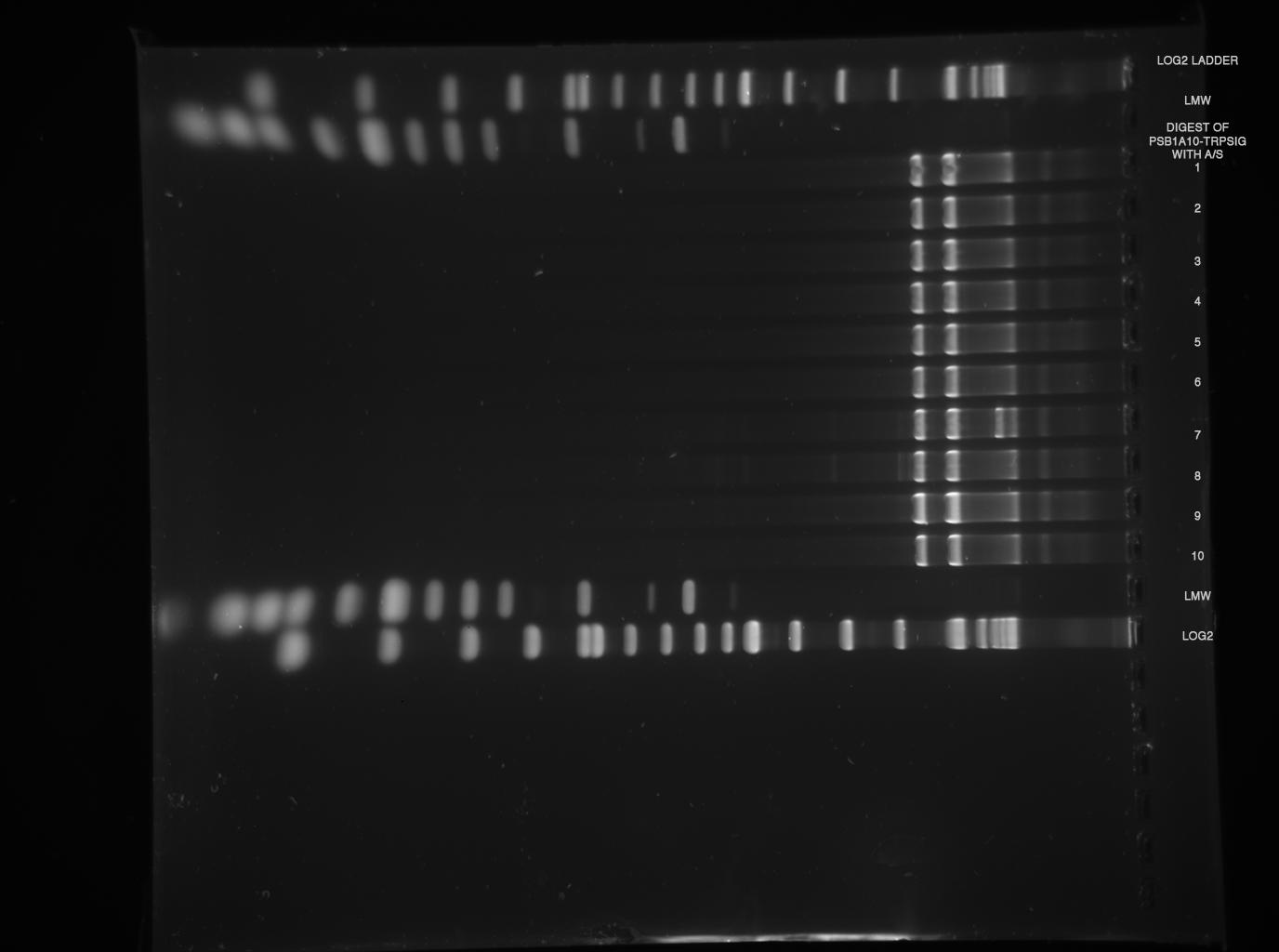 --> digestions worked, but again, no insert can be found, despite gel was at maximum resolution ( 3h 120 V, see LMW)
--> digestions worked, but again, no insert can be found, despite gel was at maximum resolution ( 3h 120 V, see LMW)
in vitro measurements
f$%&&§ s%§$! Again, nothing worked! Although we saw an increasing "GFP signal" comparable to 16.06.10, taking spectra suggested we DON'T see significant GFP-production! We used new water for preparing the samples, cleaned cuvettes with "new water", used other DNA-samples etc. Somehow, it seems as if we don't express GFP (we compared Christoph's results! We should see a really significant spectrum!
Next steps:
- Try in vivo measurements, just using psb1A10_xTerm without Signal (thus just measuring plasmid) to proof if kit or measuring plasmid causes this problem!
Close
Week12
in vivo constructs First steps Promega Kit
22.06.2010
Transformation
- psb1A10_HisTerm/TrpTerm into BL21 (DE3) RIL
- psb1A10_Monster-Trp No. 7, 8, 10 (Positiv control) into DH5-a
Liquid culture
- psb1A10_TrpSig (Positiv control) No. 2
23.06.2010
- pSBN1A10_TrpSig - 40 µL, 12.5 ng/µL -->very low amount of DNA...
- culture is slightly red? -> strange because there cannot be any rfp-insert with constitutive promoter as the construct was built up from pSB1A10_TrpTerm (digested) and TrpSignal (PCR product)
- with MonsterTrp, #7,8,10 (Carbamp)
- 50 ml cultures of BL21 (DE3) RIL
- pSB1A10_HisTerm
- pSB1A10_TrpTerm
0.2%
24.06.2010
- MonsterTrp #7,8,10
- positive Control
- concentrations are too low for sequencing --> again we have to set up 5 ml cultures for tomorrow
25.06.2010
Plasmid purification and sequencing
- Monster_Trp 7, 8, 10 and psB1A1ß_TrpSig plasmids are isolated (concentrations up to 110 ng/ul) and sent for sequencing
- psB1A1ß_TrpSig liquid culture was completely pink! still not clear what happend (wrong labeling of digested psB1A10_Trpterm?) --> wait for sequencing details
Fluorescence Measurements
- Induction by putting Arabinose directly into cuvettes with cells IS NOT WORKING at all! Expression of GFP increases, but marginally. probably, despite stirring, oxygen is lacking?
- Induction on shaker work perfectly --> both Trp and His showed strong GFP-signals, BUT: Probably, too high OD results in not exciting all GFP within the sample (incident beam is already scatterd enormously on the edge of the cuvette --> only small volume is excited correctly). For instance, a sample showing OD of 0.7 shows a signal of 30 a.u., diluted to OD 0.35 signal falls only to 19 a.u.! Thus dilution did not result in a linear decrease of flourescence! a.u. !!! We diluted down to OD 0.1; the result: OD´s smaller than 0.4 show linear change of fluorescence signal --> using OD´s up to 0.4 results in meaningful measurements!!!
Close
Week13
in vivo constructs Testing pSB1A10
28.06.2010
- psB1A10_TrpSig (Positive control) worked: Trp-Sig is inside, directly in front of RFP (without the promotor of the measurement plasmid insert [http://partsregistry.org/Part:BBa_J04450 BBa_J04450], so it seems like everything worked. Furthermore, i performed a promotor prediction with the following tool [http://linux1.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb bacteria promotor prediction tool] to proof if our Trp-Sig in combination with the flanking regions is not forming a promotor,by mischance. According to this, there are two promotors, BUT:
one in and after the suffix (so it should be in each of our constructs), but the tools says theres is no known sigma-factor for this promotor! the second one is within the RFP and there is a sigma-factor for this one ( rpoD16). So i don´t see a explanation, why our colonies were pink in contrast to the other "Messplasmids".
None of the Monsterplasmids contains the signal construct. Probably, the problem is there is no selection methods which allowed us to distinguish uncut plasmids.... --> we should discuss at our next meeting, one possiblity would be connecting our construct to a resistance marker. I summed up all sequences in this document: File:25.06.-sequenzierung.doc
- Induction in cuvette and measuring fluorescence at the same time IS NOT WORKING! (probably cells are not growing and expressing very well, maybe lacking oxygen despite stirring. Bleaching is more unlikely)
In vivo, measuring plasmid (at least GFP) works! we optimized the paramters for fluorescence measurment! We tried different OD´s and found out that only measurments below OD 0.4 result in meaningful measurements.
as a result, in vitro expression did somehow not work, reasons are unclear, maybe too low DNA-concentrations.
positvie control psB1A10_TrpSig was pink again, we have to wait the results from GATC
29.06.2010
Close
Week14
in vivo constructs Invitrogen Kit
01.07.2010
- using Invitrogen kit
- measuring kinetics for 3 h @ 37°C
- 40 µL + 5 µL pSB1A10_TrpSig (126 ng/µL) + 0.5 µL 100x L-Arabinose (=0.2%) + 4.5 µL H2O
- observations: GFP signal grows, after 30 min it crashes. RFP grows
- emission spectra for GFP and RFP result in no spectrum
- looks strange, a problem might be evaporation of liquid and hence scattering of light which produces artefacts
- pSB1A10_TrpSig (DH5a), 5 ml for miniprep
- pSB1A10 XS (DH5a), 5 ml for miniprep
02.07.2010
- pSB1A10_XS: 30 µL 10 ng/µL
- pSB1A10_TrpSig: 30 µL 10 ng/µL
- BL21 with pSB1A10_XS (positive control without insert and no without any bio brick site left)
Close
Week15
Testing pSB1A10
05.07.2010
- 250 ml of DH5a pSB1A10_XS
- 20 ml BL21 pSB1A10_XS
- in vitro transcription measurement planned
- check In_vitro_Measurements
06.07.2010
- BL21 pSB1A10_XS - positive control (to check the measurement plasmid...)
- GFP, RFP Fluorescence
- induced with 0.2% arabinose in (1), uninduced (2)
- at OD 0.15: GFP/RFP emissions spectra /100706/spectra/gfp10 and rfp10
- 2.5 h kinetic measurement GFP/RFP /100706/kinetics/
- OD 0.7 (1) and 0.64 (2) after 2.5 h --> GFP/RFP emissions spectra /100706/spectra/gfp11,rfp11,gfp21,rfp21
- in addition for 4 h a culture at OD 0.8 induced (with 0.2% Arab), spectra taken afterwards at 1:15 dilution (OD 0.39) gpf_ku and rfp_ku
- observation: measurement plasmid is totale verarsche. RFP is not expressed at all, or this protein is not rfp. whatever.
Close
Week16
The Era of Exams
No Lab work this week, everybody is busy studying for their exams...
Close
Week17
The Era of Exams
No Lab work this week, everybody is busy studying for their exams...
Close
Week18
The Era of Exams
No Lab work this week, everybody is busy studying for their exams...
Close
Week19
The Era of Exams
No Lab work this week, everybody is busy studying for their exams...
Close
Week20
Construction of new Measurement Plasmid
09.08.2010
- overnight cultures inoculated from Glycerolstock J06702(mCherry generator) in pSB1A2 from Christoph.
10.08.2010
- MiniPrep of pSB1A2-mCherry using ZymoKit
- Digestion
- pSB1A2-mCherry E/P
- pSB1A2-mCherry X/P
- pSB1A10-HisTerm S/P
- pSB1A10-TrpTerm S/P
- pSB1A10-HisTerm E/P
- Purified with agarose gel (1%)
- Gel Doc broken => no picture
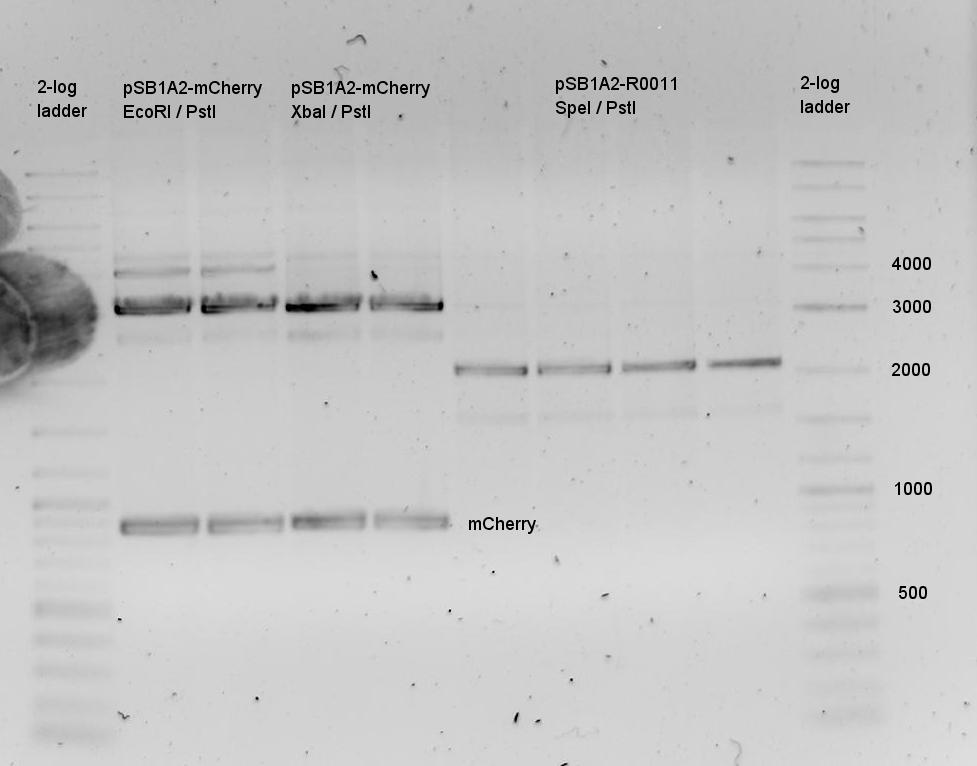
- Description: mCherry cut was ok, Plasmid was cut at least once (linear DNA), generally contaminated with genomic DNA
- Ligation
- 50 ng Plasmid and 34 ng Insert
- ca. 30min @ RT
- Transformation of DH5a cells with ligation samples
(=> no colonies the next day)
- overnight cultures
- pSB1A2-mCherry from Christoph`s stock
- pSB1A10-HisTerm from earlier plate
- pSB1A10-TrpTerm from earlier plate
(=> pSB1A10-TrpTerm and pSB1A10-HisTerm did not grow until next day)
11.08.2010
- MiniPrep of pSB1A2-mCherry using ZymoKit
- analytic gel of Mini preps and ligation of the previous day
- preps still hold genomic DNA
- mCherry Plasmid runs at ca. 2400 bp
- Digestion
- pSB1A2-mCherry E/P
- pSB1A2-mCherry X/P
- Purified with agarose gel (1%)
- Gel Doc broken => no picture
- Description: mCherry cut was ok, stil contaminated with genomic DNA
- Ligation
- Plasmid (from previous day) and mCherry-Insert
- ca. 30 min @ RT
- Transformation of DH5a cells with
- ligation samples (=> no colonies the next day)
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
- overnight cultures
- pSB1A2-mCherry from Christoph`s stock
12.08.2010
- MiniPrep of pSB1A2-mCherry using ZymoKit
- analytic gel of Mini preps and ligation of the previous day
- preps still hold genomic DNA
- mCherry Plasmid runs at ca. 2400 bp
- Gel (1%)
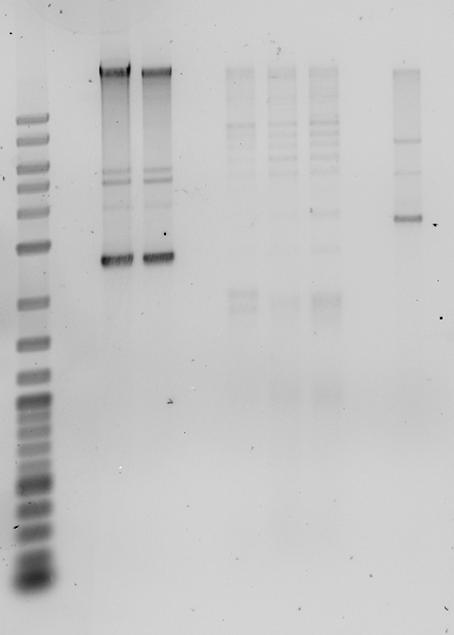
- Digestion
- pSB1A2-mCherry X/P
- pSB1A10-HisTerm S/P
- pSB1A10-TrpTerm S/P
- Purified with agarose gel (1%)
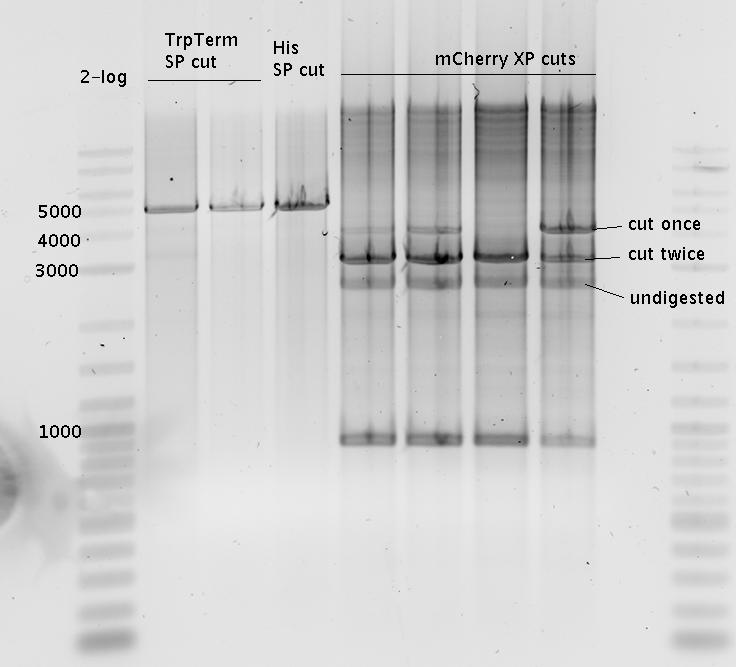
- Ligation
- Plasmid and mCherry-Insert
- ca. 30 min @ RT
- Transformation of DH5a cells with
- ligation samples (=> no colonies the next day)
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
- overnight cultures
- pSB1A2-mCherry from Christoph`s stock
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
Caution: ran out of gas => not steril?
13.08.2010
- MiniPrep using ZymoKit
- pSB1A2-mCherry
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
- analytic gel of Mini preps and ligation of the previous day
- low concentration
- Gel (1%)
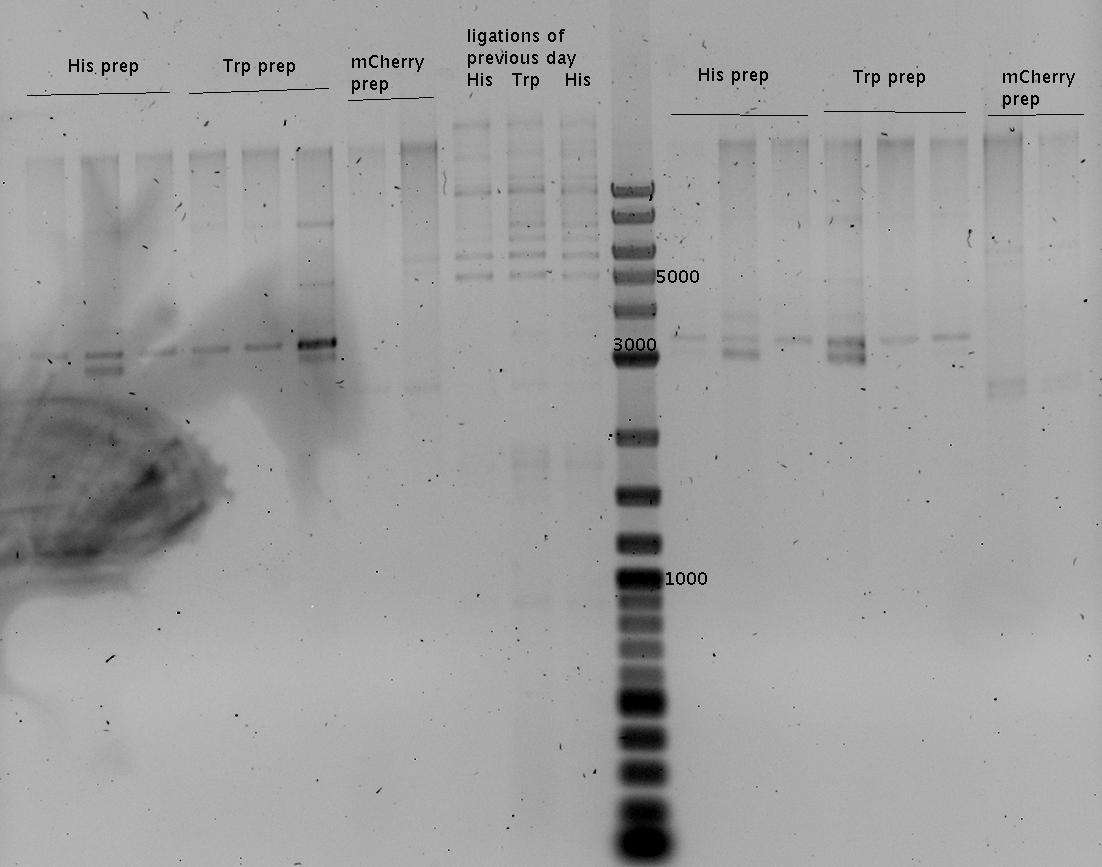
Close
Week21
Construction of new Measurement Plasmid
16.08.2010
- Concentrating MiniPrep-Samples using ZymoKit
- pSB1A2-mCherry
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
- Digestion
- pSB1A2-mCherry E/P
- pSB1A2-mCherry X/P
- Purified with agarose gel (1%)
 => no mCherry band!!!
=> no mCherry band!!!
- Purification using Zymo Concentrator Kit
- pSB1A10-HisTerm S/P
- pSB1A10-TrpTerm S/P
- Ligation
- Plasmid (from earlier date) and mCherry-Insert (from 11.08)
- ca. 30 min @ RT
- used new ligase and new ligase buffer
- Transformation of DH5a cells with
- ligation samples (=> colonies found the next day)
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
- pSB1A10-RFP (BioBrick Standard)
- overnight cultures
- pSB1A2-mCherry from Christoph`s stock
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
17.08.2010
- MiniPrep using ZymoKit
- pSB1A2-mCherry
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
- Digestion
- pSB1A2-mCherry X/P
- pSB1A10-HisTerm S/P
- pSB1A10-TrpTerm S/P
- => heating block went up to 50°C
- Purified "digestion" samples with ZymoKit => stored for next day
- Picked 12 colonies from previous day's ligation
- => Colony PCR => Gel (2%)
- Purified with agarose gel (1%)
- Gel Doc broken => no picture
- Description: mCherry cut was ok, stil contaminated with genomic DNA
- Ligation
- Plasmid (from previous day) and mCherry-Insert
- ca. 30 min @ RT
- Transformation of DH5a cells with
- ligation samples (=> no colonies the next day)
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
- overnight cultures
- pSB1A10-RFP (plate from previous day)
- pSB1A2-mCherry
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
PROBLEM:
mCherry has a SgrA1-cleavage site! These constructs cannot be used. Starting all over, cloning the linker sequence first...
18.08.2010
- MiniPrep using ZymoKit
- pSB1A2-mCherry
- pSB1A10-HisTerm
- pSB1A10-TrpTerm
- pSB1A10-RFP
- Analytical agarose gel (1%):
- Digestion
- pSB1A10-HisTerm SgrAI/PstI
- pSB1A10-TrpTerm SgrAI/PstI
- pSB1A10-RFP SgrAI/PstI
- =>RFP has a SrgAI cleaving site. Discarded RFP digestion.
- preparativ agarose gel (1%):
- Soubilization of SrgAI-PstI Linker
- Ligation
- Plasmid His-Term(Trp-Term) and Linker
- ca. 30 min @ RT
- Transformation of DH5a cells with
- overnight cultures
- pSB1A2-mCherry
- pSB1A10-TrpTerm
19.08.2010
- MiniPrep using ZymoKit
- analytic agarose gel (1%) from various mCherry Preps
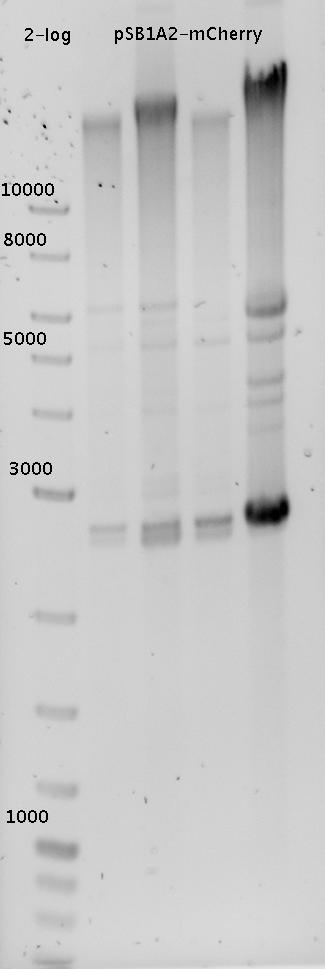
- Picked 6 colonies from pSB1A10-HisTerm-linker and pSB1A10-TrpTerm-linker each (previous day's ligation)
- => Colony PCR => Gel (1.5%)
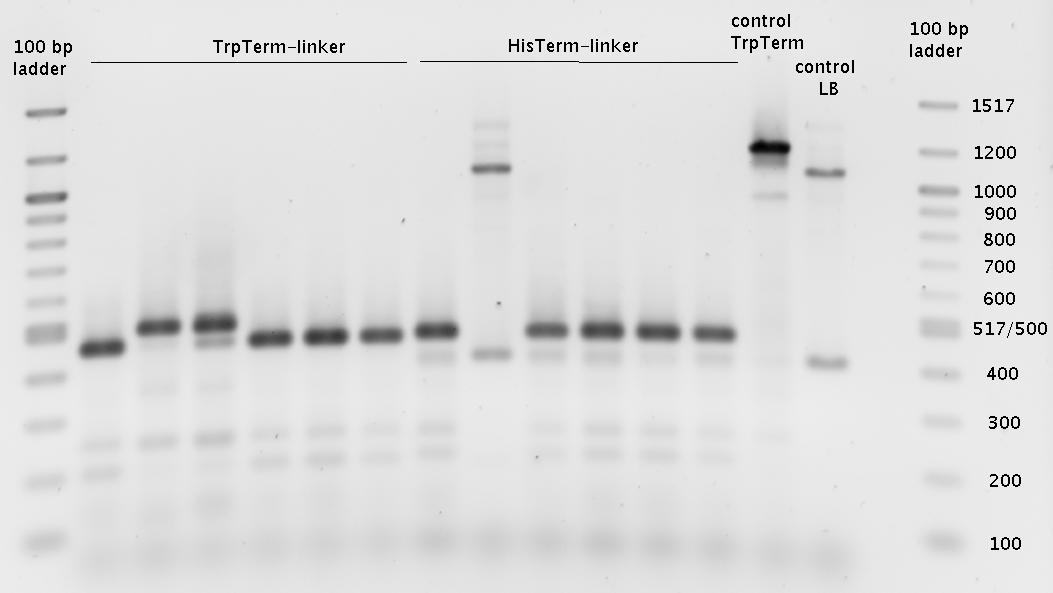
- overnight cultures (600µl)
- pSB1A2-mCherry
- pSB1A2-R0011
- pSB1A10-HisTerm-linker (#7, 11, 12)
- pSB1A10-TrpTerm-linker (#1, 2, 4)
20.08.2010
- MiniPrep using ZymoKit
- pSB1A2-mCherry
- pSB1A2-R0011
- pSB1A10-TrpLinker (picked Colonies)
- pSB1A10-HisLinker (picked Colonies)
- analytical Digestion
- pSB1A10-HisLinker SgrAI /EcoRI
- pSB1A10-HisLinker NsiI
- pSB1A10-TrpLinker SgrAI /EcoRI
- analytical agarose gel (1.5%)
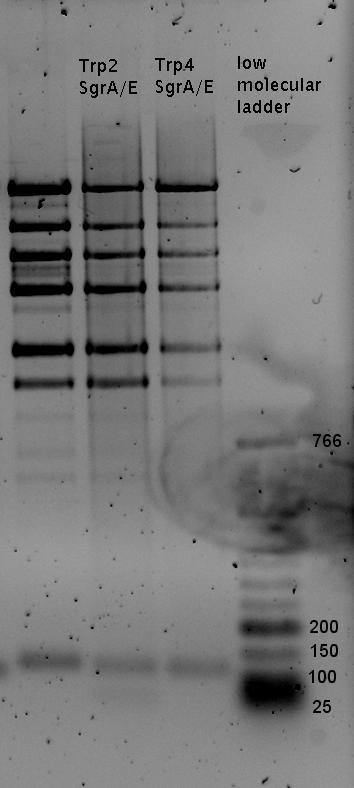
- preparativ Digestions
- pSB1A2-mCherry EcoRI /PstI
- pSB1A2-mCherry XbaI /PstI
- pSB1A2-R0011 SpeI /PstI
- pSB1A10-HisLinker SpeI /PstI
- pSB1A10-HisLinker EcoRI /PstI
- pSB1A10-TrpLinker SpeI /PstI
- pSB1A10-TrpLinker EcoRI /PstI
- PCR_BB1006 XbaI /PstI
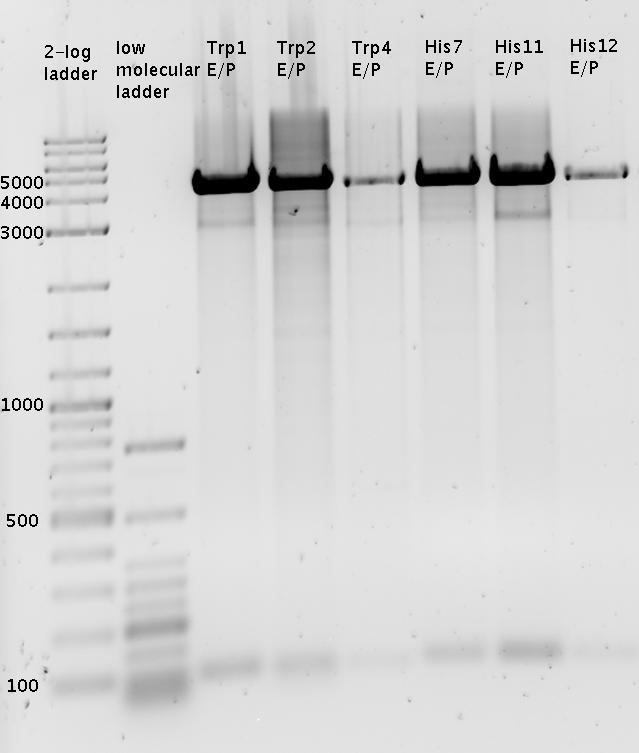
Close
Week22
Construction of new Measurement Plasmid
23.08.2010
- Ligation
- pSB1A10-HisLinker SpeI /PstI + mCherry XbaI /PstI
- pSB1A10-HisLinker EcoRI /PstI + mCherry EcoRI /PstI
- pSB1A10-TrpLinker SpeI /PstI + mCherry XbaI /PstI
- pSB1A10-TrpLinker EcoRI /PstI + mCherry EcoRI /PstI
- pSB1A2-R0011 SpeI /PstI + PCR_BB1006 XbaI /PstI
- Transformation of DH5a cells
24.08.2010
- Picked 2 colonies per plate (previous day's ligation)
- R0011_B1006Sig
- Trp1_mCherry
- Trp2_mCherry
- His7_mCherry
- His11_mCherry
- pSB1A10_mCherry
- Colony PCR of picked colonies with prefix/suffix primers
- analytical agarose gel 1 (1.5%)
- analytical agarose gel 1 (1.5%)
Trp=R0011, R0011= Trp :)
Faint bands at the correct length can be guessed.
- overnight cultures (5 ml)
- pSB1A10_TrpTerm_mCherry_linker
- pSB1A10_HisTerm_mCherry_linker
- pSB1A10_mCherry_linker
25.08.2010
- Miniprep using Zymo Miniprep-Classic Kit:
- pSB1A10_TrpTerm_mCherry_linker
- pSB1A10_HisTerm_mCherry_linker
- pSB1A10_mCherry_linker
- analytical digestions
- pSB1A10_TrpTerm_mCherry_linker EcoRI /PstI
- pSB1A10_HisTerm_mCherry_linker EcoRI /PstI
- pSB1A10_mCherry_linker EcoRI /PstI
- analytical agarose gel 1 (1.0%)
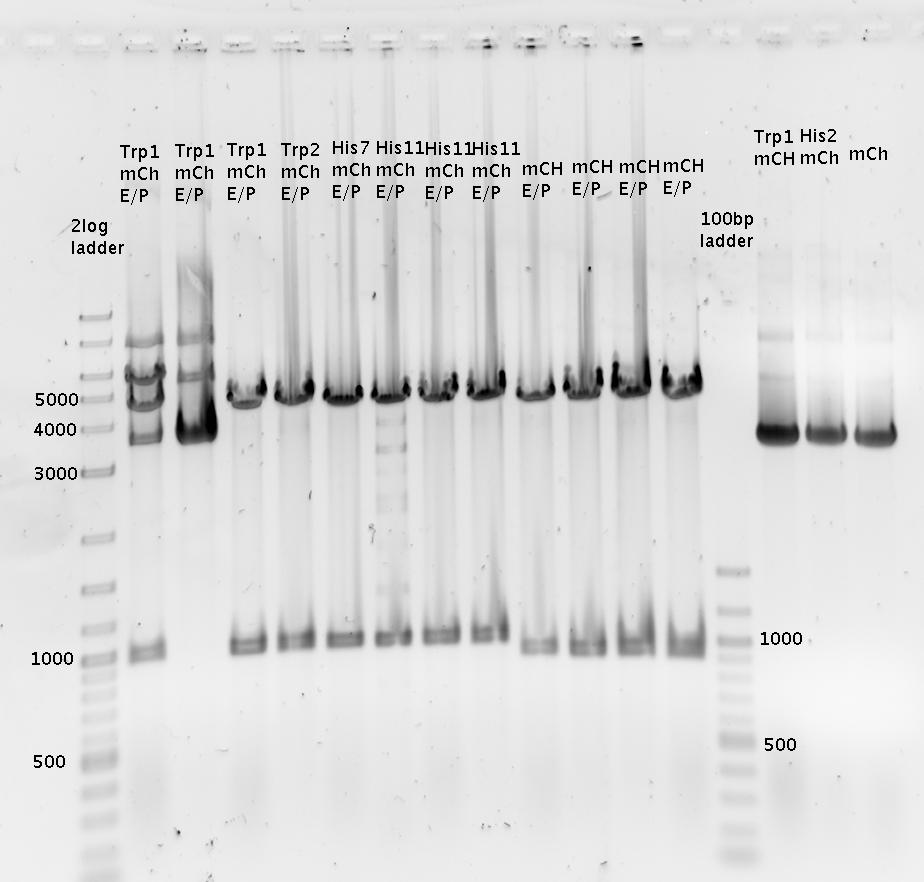
- Picked 2 colonies per plate (day before yesterday's ligation)
- R0011_B1006Sig
- Trp1_mCherry
- Trp2_mCherry
- His7_mCherry
- His11_mCherry
- pSB1A10_mCherry
- Colony PCR of picked colonies with prefix/suffix primers
- analytical agarose gel 1 (1.5%)
- analytical agarose gel 1 (1.5%)
- overnight cultures (5 ml)
- pSB1A10_TrpTerm_mCherry_linker
- pSB1A10_HisTerm_mCherry_linker
- pSB1A10_mCherry_linker
26.08.2010
- Miniprep using Zymo Miniprep-Classic Kit:
- pSB1A10_TrpTerm_mCherry_linker
- pSB1A10_HisTerm_mCherry_linker
- pSB1A10_mCherry_linker
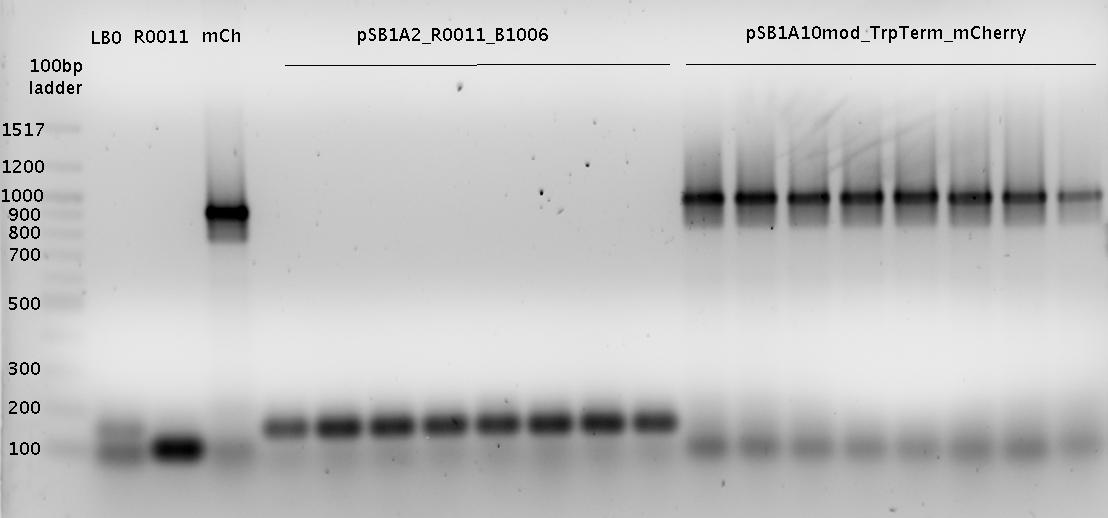
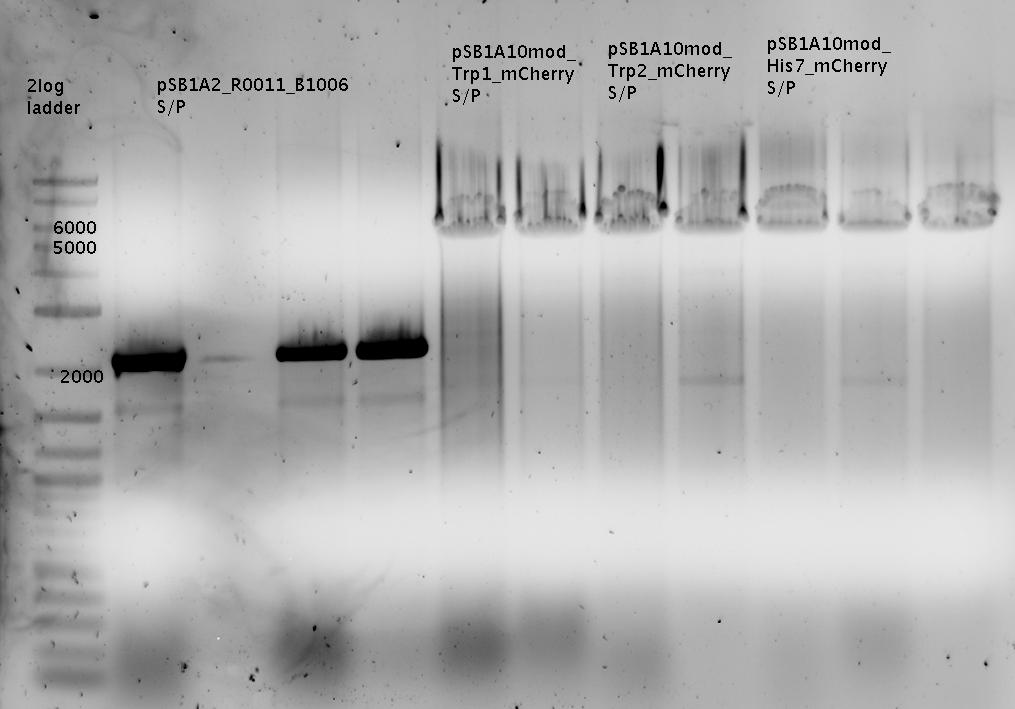
- pSB1A2_R0011_B1006
- PCR
- Trp-Signal (R0011_Sig_B0014)
- His-Signal (R0011_Sig_B0014)
- Terminator B0014
- analytical digestions
- pSB1A10_mCherry_linker EcoRI /PstI
- preparative digestion
- pSB1A10_TrpTerm_mCherry_linker SpeI/PstI
- pSB1A10_HisTerm_mCherry_linker SpeI/PstI
- pSB1A2_R0011_B1006 SpeI/PstI
- PCR Trp-Sig XbaI/PstI
- PCR His-Sig XbaI/PstI
- B0014 XbaI/PstI
- preparative agarose gel 1 (1.0%)
 last lane: pSB1A10_His11_mCherry SpeI/PstI
last lane: pSB1A10_His11_mCherry SpeI/PstI
- analytical agarose gel (1.0 %)
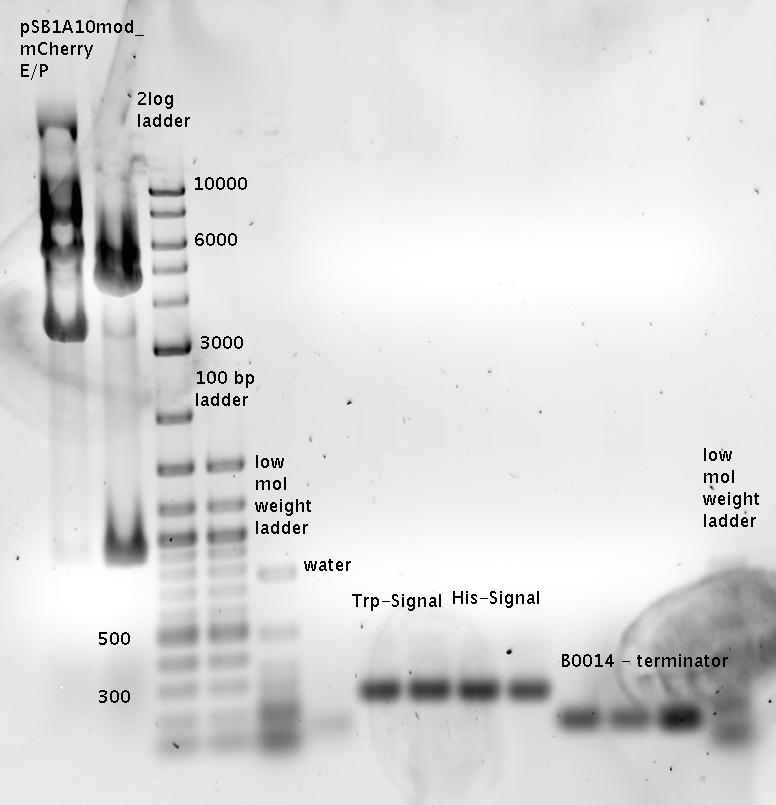
- Ligations
- pSB1A10_TrpTerm_mCherry_linker + Trp-Signal (R0011_Sig_B0014)
- pSB1A10_HisTerm_mCherry_linker + His-Signal (R0011_Sig_B0014)
- pSB1A2_R0011_B1006 + Terminator B0014
- Transformation of Ligation product in DH5alpha cells
- Transformation of pSB1A10_mCherry_linker in BL21
27.08.2010
for sequencing
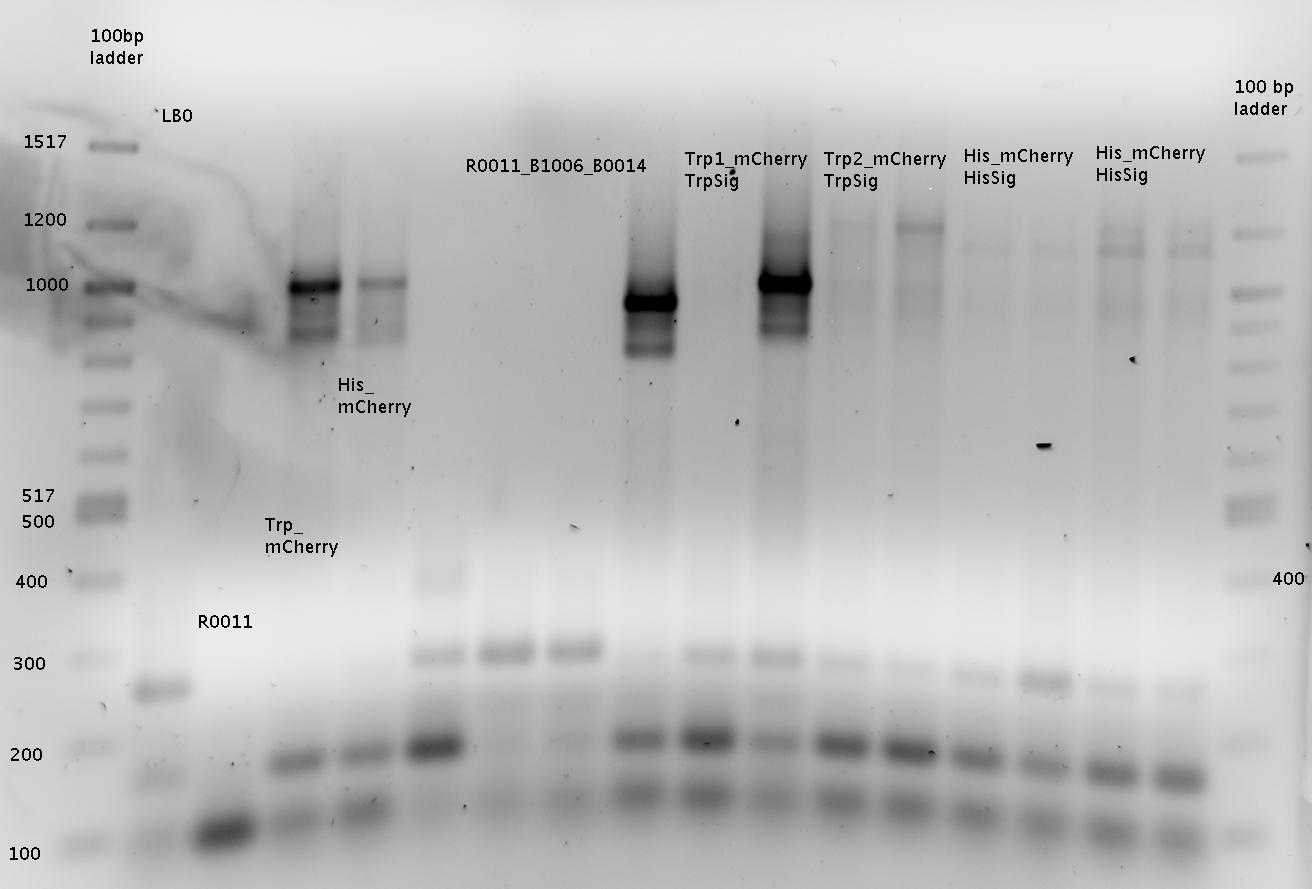 R0011=R0011_B1006!!!
R0011=R0011_B1006!!!
- Sequencing
- pSB1A2_R0011_B1006 4b with primer Biobrick VR
- pSB1A10mod_mCherry 27b with primer GFP_FP and Biobrick VR
- pSB1A10mod_mCherry 32a with primer GFP_FP and Biobrick VR
Close
Week23
Construction of new Measurement Plasmid Testing new Measurement Plasmid
30.08.2010
- Colony PCR
- picked two colonies per plate from 26.08' ligation
- Program: colonypcr, modified elongation time: 1.15 instead of 1.00
- analytical agarose gel (1.5%)
Gel 1:
=> samples named "1x" to "8x" for pSB1A2-R0011-BB1006Sig-B0014 colonies
=> samples named "9x" to "12x" for pSB1A10mod-TrpTerm-mCherry-TrpSig colonies
Gel 2:
=> samples named "13x" to "16x" for pSB1A10mod-TrpTerm-mCherry-TrpSig colonies
=> samples named "17x" to "24x" for pSB1A10mod-HisTerm-mCherry-HisSig colonies
Interpretation for Gel1 and Gel2:
Ligation worked for the samples 1x-6x (pSB1A2-R0011-BB1006Sig-B0014), 9x-16x (pSB1A10mod-TrpTerm-mCherry-TrpSig), 18x-24x (pSB1A10mod-HisTerm-mCherry-HisSig)
- over night cultures:
- pSB1A10mod_HisTerm_mCherry_HisSignal
- pSB1A10mod_TrpTerm_mCherry_TrpSignal
- PSB1A2_R0011_BB1006_B0014
- Received sequencing results from GATC. All Sequences are okay:
- pSB1A10mod_mCherry (27b) and (32a)
- pSB1A2_R0011_BB1006 (4b)
31.08.2010
- Fluorescence measurement (positive control experiment):
- Settings: GFP-Excitation: 501 nm; mCherry-Excitation: 587 nm;
- endpoint measurements of:
- Timepoints of measurement: 3 h after induction and 9 h after induction (1.5 h and 7 h for 15x-sample)
- Samples:
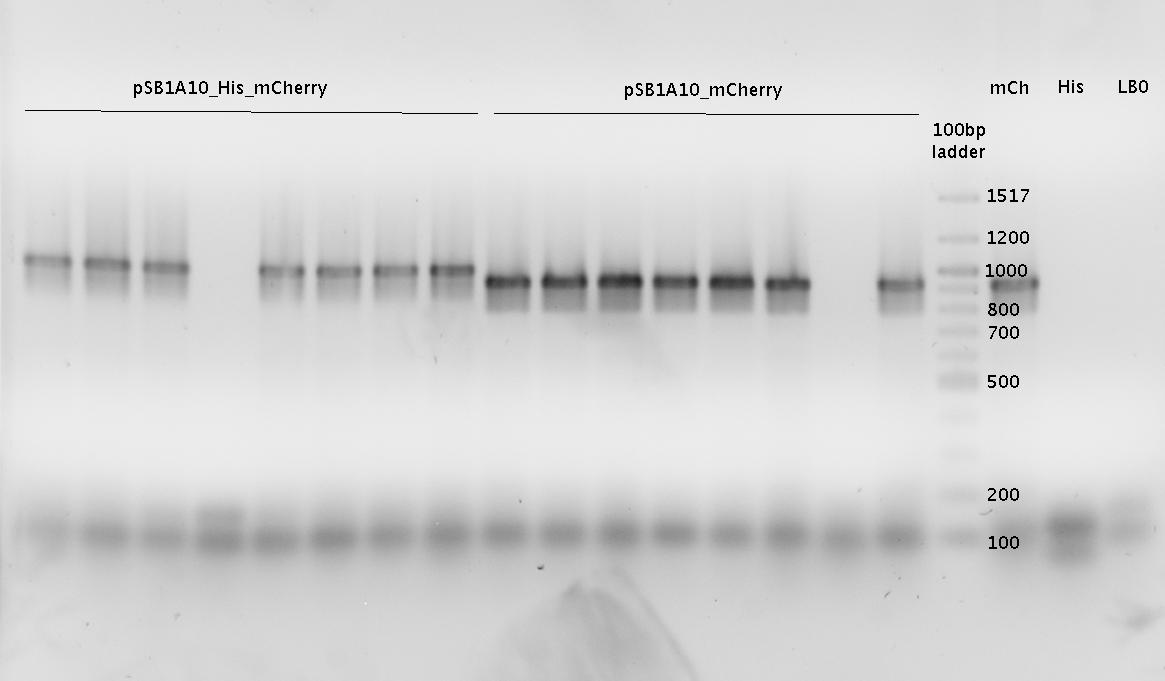
- pSB1A10mod-mCherry (27b) in BL21 cells, induced with ca. 0.4% L-Arabinose
- pSB1A10mod-mCherry (27b) in BL21 cells, not induced
- kinetic measurement of induced (0.4% L-Arabinose) BL21 cells carrying pSB1A10mod-mCherry (27b)
- Results:
- NO mCherry signal detected at all: The GFP signal shows a nice and strong increase; the RFP channel did not change at all.
- GFP signal looks perfect: strong if induced, neglectable if not!
- => System seems not capable of serving as a testing system for our switches!
- Glycerol stocks
- in DH5a cells:
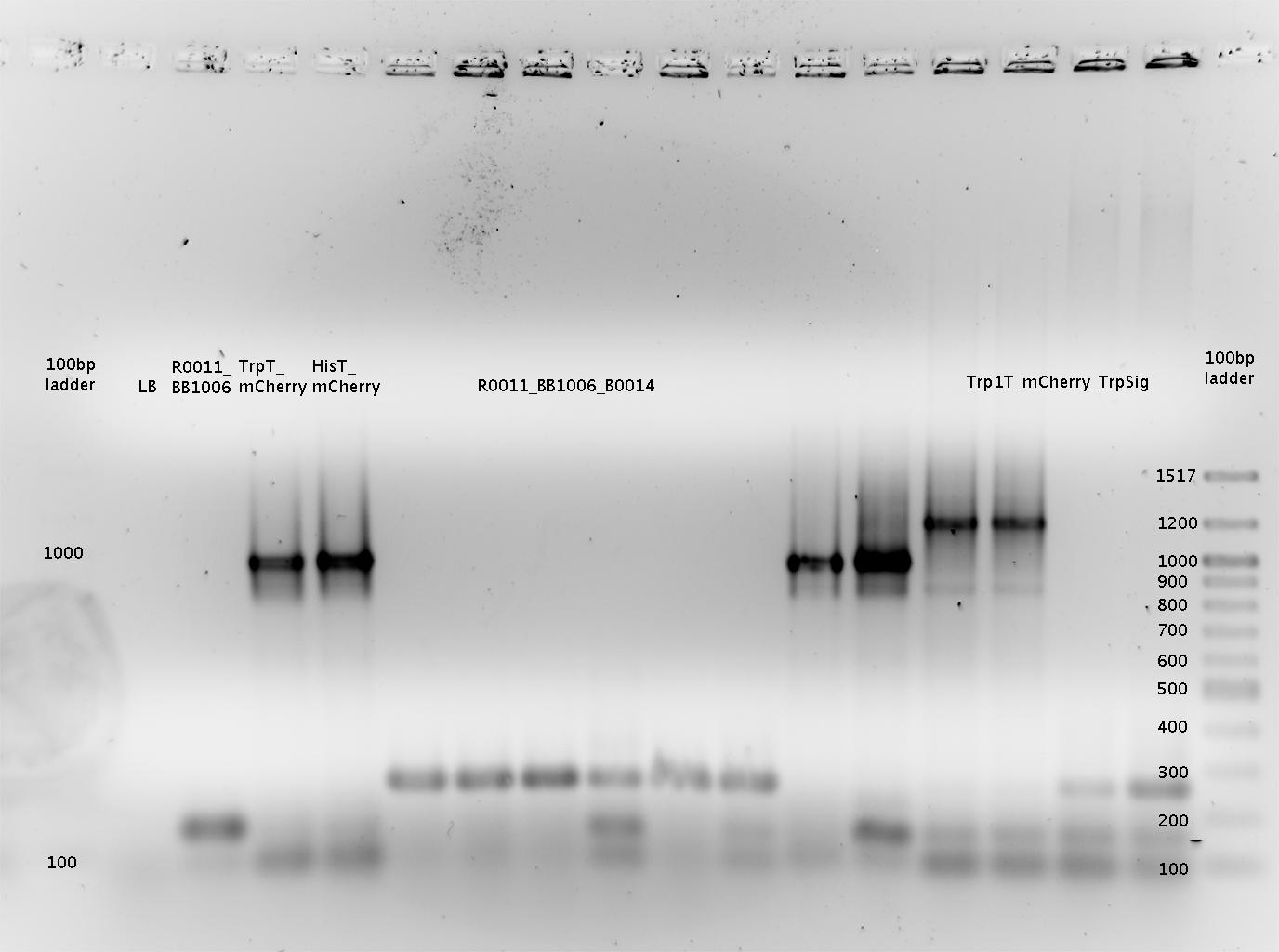
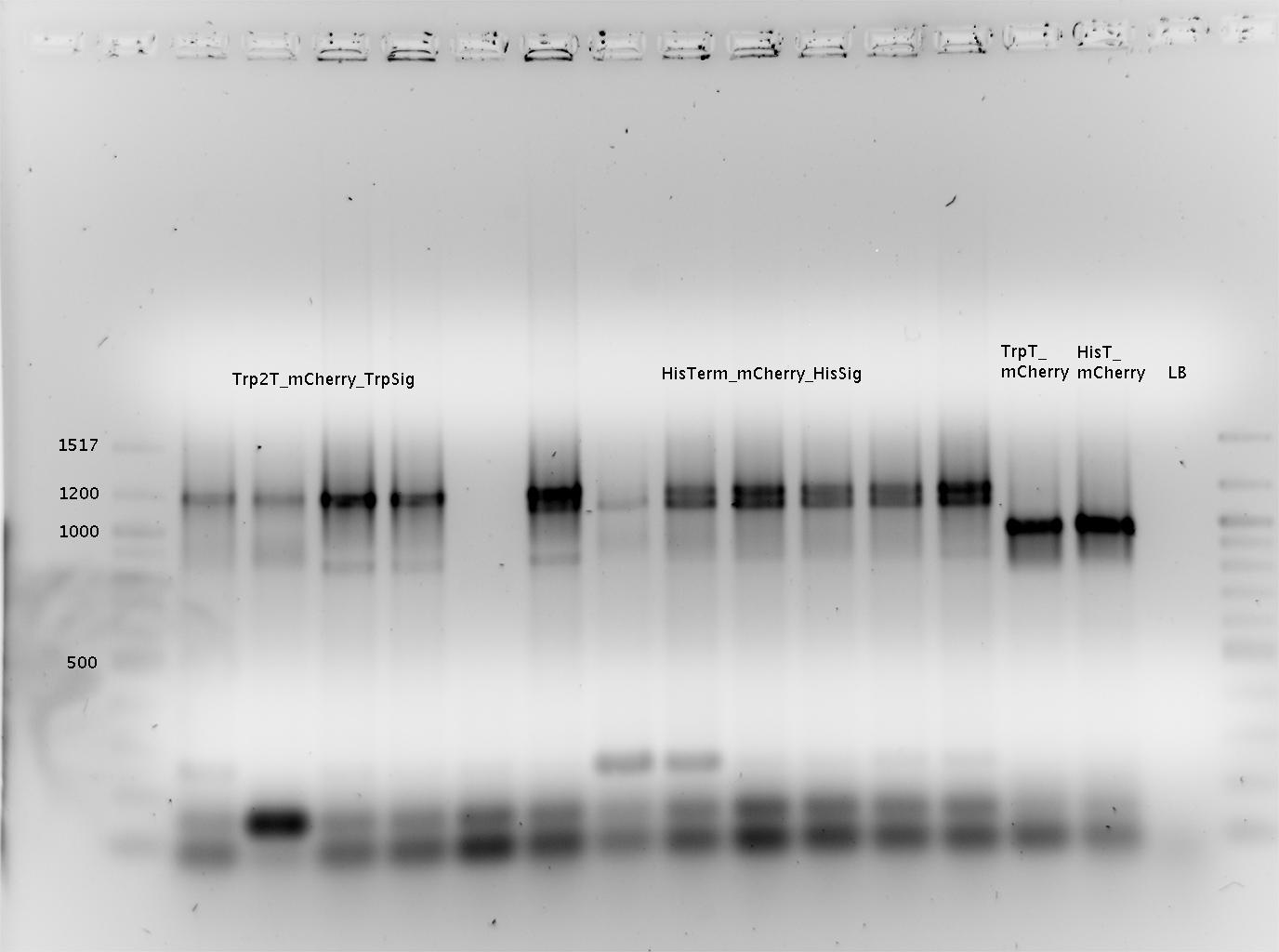
- pSB1A10mod-TrpTerm-mCherry-TrpSig (9x)
- pSB1A10mod-TrpTerm-mCherry-TrpSig (15x)
- pSB1A10mod-TrpTerm-mCherry-TrpSig (10x) (sequence verified)
- pSB1A10mod-HisTerm-mCherry-HisSig (15x)
- pSB1A10mod-HisTerm-mCherry-HisSig (18x)
- pSB1A10mod-HisTerm-mCherry-HisSig (23x) (sequence verified)
- pSB1A10mod-mCherry (32a) (sequence verified)
- pSB1A10mod-mCherry (27b) (sequence verified)
- pSB1A2mod R0011-BB1006Sig-B0014 (2x) (sequence verified)
- pSB1A2mod R0011-BB1006Sig-B0014 (3x)
- pSB1A2mod R0011-BB1006Sig (4b) (sequence verified)
- in BL21 cells:
- pSB1A10mod-mCherry (27b) (sequence verified)
- "x" refers to Colony-PCR of 30.08.2010
- 5ml Over night cultures
- pSB1A10mod-TrpTerm-mCherry-TrpSig (9x, 15x, 10x)
- pSB1A10mod-HisTerm-mCherry-HisSig (18x, 23x, 15x)
- pSB1A2mod R0011-BB1006Sig-B0014 (2x, 3x)
01.09.2010
- MiniPrep using Zymo classical kit. Samples:
- pSB1A10mod-TrpTerm-mCherry-TrpSig (9x, 15x, 10x)
- pSB1A10mod-HisTerm-mCherry-HisSig (18x, 23x, 15x)
- pSB1A2mod R0011-BB1006Sig-B0014 (2x, 3x)
- Fluorescence measurement (positive control experiment):
- endpoint measurements:
- Timepoints of measurement: 3 h after induction and 9 h after induction (1.5 h and 7 h for 15x-smaple)
- Settings: GFP-Excitation: 501 nm; mCherry-Excitation: 587 nm; RFP-Excitation: 584 nm
- Samples:
- pSB1A10mod-mCherry (32a) in DH5a cells, induced with ca. 0.4% L-Arabinose
- pSB1A10mod-mCherry (32a) in DH5a cells, not induced
- pSB1A10mod-mCherry (32a) in BL21 DE3 cells, induced with ca. 0.4% L-Arabinose
- pSB1A10mod-mCherry (32a) in BL21 DE3 cells, not induced
- pSB1A10-RFP, in DH5a cells, induced with ca. 0.4% L-Arabinose
- pSB1A10-RFP, in DH5a cells, not induced
- pSB1A10mod-TrpTerm-mCherry-TrpSig (15x), in DH5a cells, induced with ca. 0.4% L-Arabinose
- pSB1A10mod-TrpTerm-mCherry-TrpSig (15x), in DH5a cells, not induced
- Results:
- Very strong RFP signal in pSB1A10-RFP, induced and not induced
- For the first time we saw a weak but easily-detectable mCherry signal in positive control samples (pSB1A10mod-mCherry) 3 hours after induction! There was hardly no difference between the uninduced and the induced control samples for mCherry. The GFP signals was strong for induced control experiments and very weak for not induced samples! The pSB1A10mod-TrpTerm-mCherry-TrpSig sample also showd a small mCHerry signal.
- After 9 hours the mCherry signals were generally reduced, whereas the GFP signals were still high in all induced samples and low in all uninduced samples.
- => Although we saw mCherry for the first time (!), the signal is to weak not reproducable! As a consequence the system can not be used to serve as a measure for our switches! Furthermore the settings of the fluorometer are ok, since we saw strong RFP signal.
02.09.2010
Starting the cloning of pBAD (BioBrick I13453) downstream of GFP
- Amplifing the Arabinose-inducable promotor pBAD
- Resuspending the BioBrick I13453 with 10 µl in well 1F in the 2010 Distribution
- PCR using 1 µl template (programm "igempcr")
- Purified using DNA Concentrator (ZymoKit)
- Digestion (EcoRI and SpeI) of PCR product and heat inactivation (20 min @ 80°C)
- Digestion of the target vectors using EcoRI and XbaI
- Samples:
- pSB1A10mod-TrpTerm-mCherry-TrpSig (9x, 10x)
- pSB1A10mod-HisTerm-mCherry-HisSig (23x, 24x)
- pSB1A10mod-mCherry (32a, 27b)
- pSB1A10mod-TrpTerm-mCherry (10b, 13b)
- pSB1A10mod-HisTerm-mCherry (18b, 24b)
- Purified using 1% agarose gel:
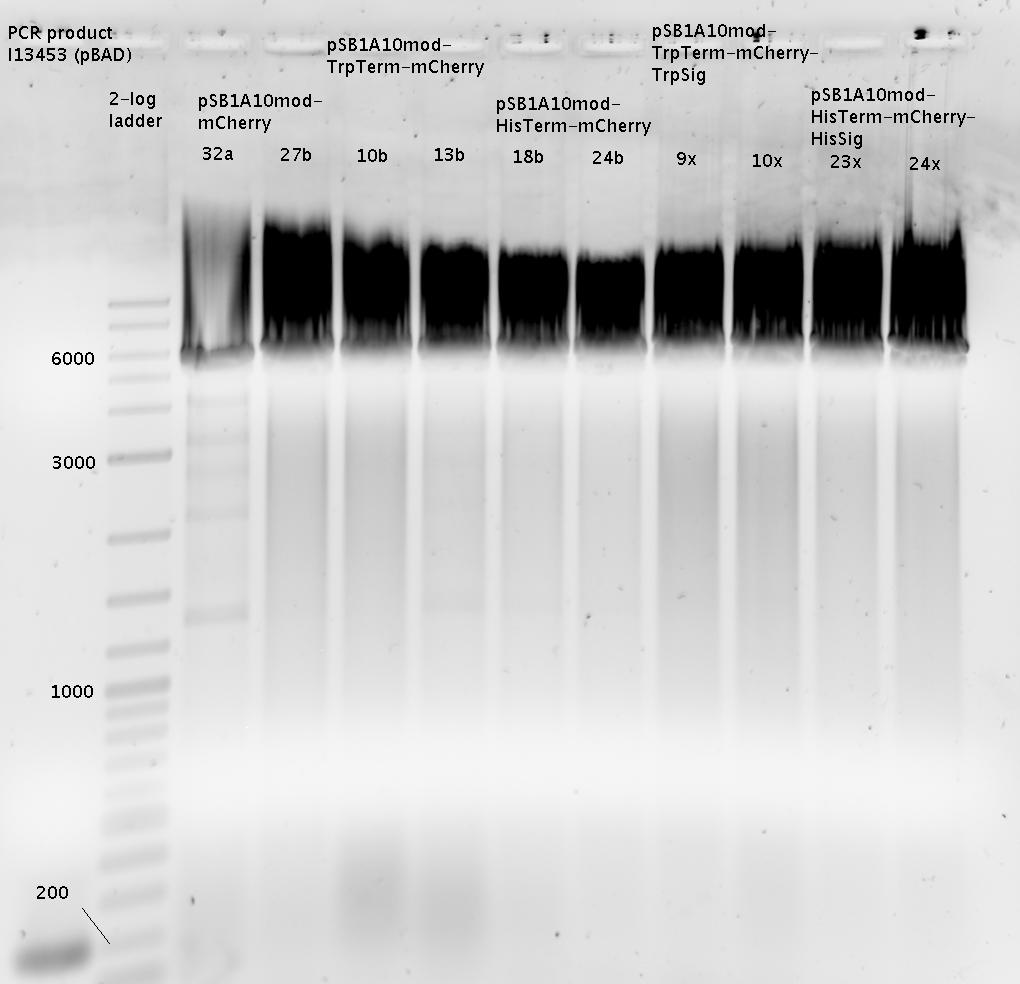
- => Interpretation: Gel is overloaded! However digestion seemed to work since the bands show correct masses.
- Extraction of bands at ca. 6000 bp
- 10 µl ligation of 50 ng of each digested vector with 8ng insert
- Transformation of DH5a cells using 8 µl ligation sample
03.09.2010
- Colony PCR
- picked two colonies per plate from 02.09' ligation
- Note: no PCR because Thermocycler was occupied
Close
Week24
in vivo constructs Testing HisTrp
06.09.2010
- Colony PCR
- using colonies picked on 03.09.
- analytical agarose gel (1.5%)
- K1= pSB1A10mod_HisTerm_mCherry_HisSig
- K2 = pSB1A10mod_TrpTerm_mCherry_TrpSig
- 5ml over night cultures:
- positive control pSB1A10mod_Pbad_mCherry
- negative His control pSB1A10mod_Pbad_HisTerm_mCherry
- His-Switch pSB1A10mod_Pbad_HisTerm_mCherry_HisSig
- Trp-Switch pSB1A10mod_Pbad_TrpTerm_mCherry_TrpSig
07.09.2010
- MiniPrep using Zymo Classical kit
- pSB1A10mod_Pbad_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry_HisSig
- pSB1A10mod_Pbad_TrpTerm_mCherry_TrpSig
- Fluorescence measurements
- Induction with 0.2% L-arabinose
- Measurement of OD600
- Fluorescence measurement at 30 min, 150 min (only Pos.Control) and 4.5 h
- Samples:
- pSB1A10mod_Pbad_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry_HisSig
- pSB1A10mod_Pbad_TrpTerm_mCherry_TrpSig
- Transformation of BL21 (DE3)
- pSB1A10mod_Pbad_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry_HisSig
- pSB1A10mod_Pbad_TrpTerm_mCherry_TrpSig
08.09.2010
- 30%Glycerol in LB_Carb
- pSB1A10mod_Pbad_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry_HisSig
- pSB1A10mod_Pbad_TrpTerm_mCherry_TrpSig
- Fluorescence measurements
- Induction with 0.2% L-arabinose
- Measurement of OD600
- Fluorescence measurement at 24 h and different OD600
- Samples:
- pSB1A10mod_Pbad_mCherry
-->OD600 0.05 reasonable for our cell measurements. Positive control works fine after 24 h induction.
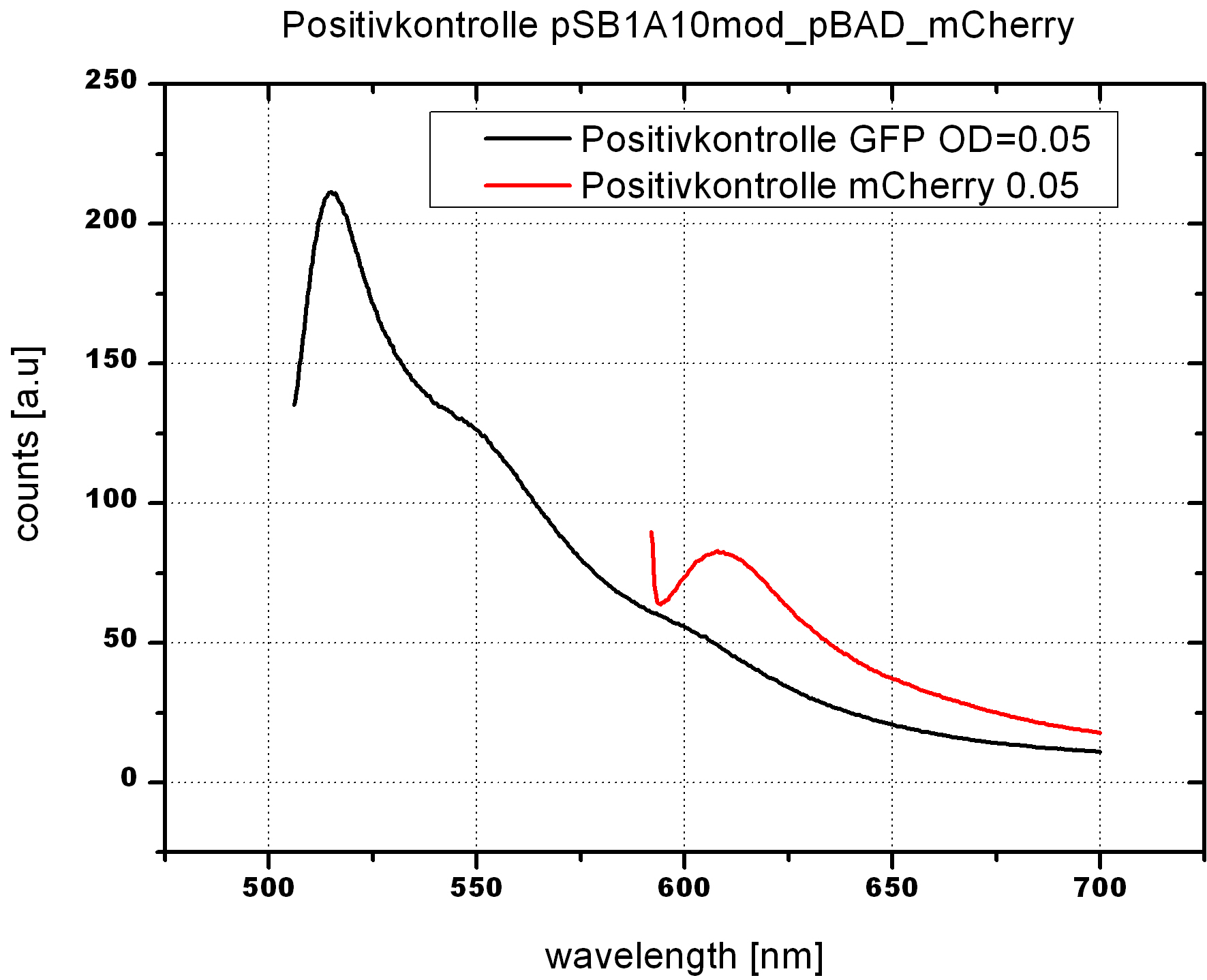
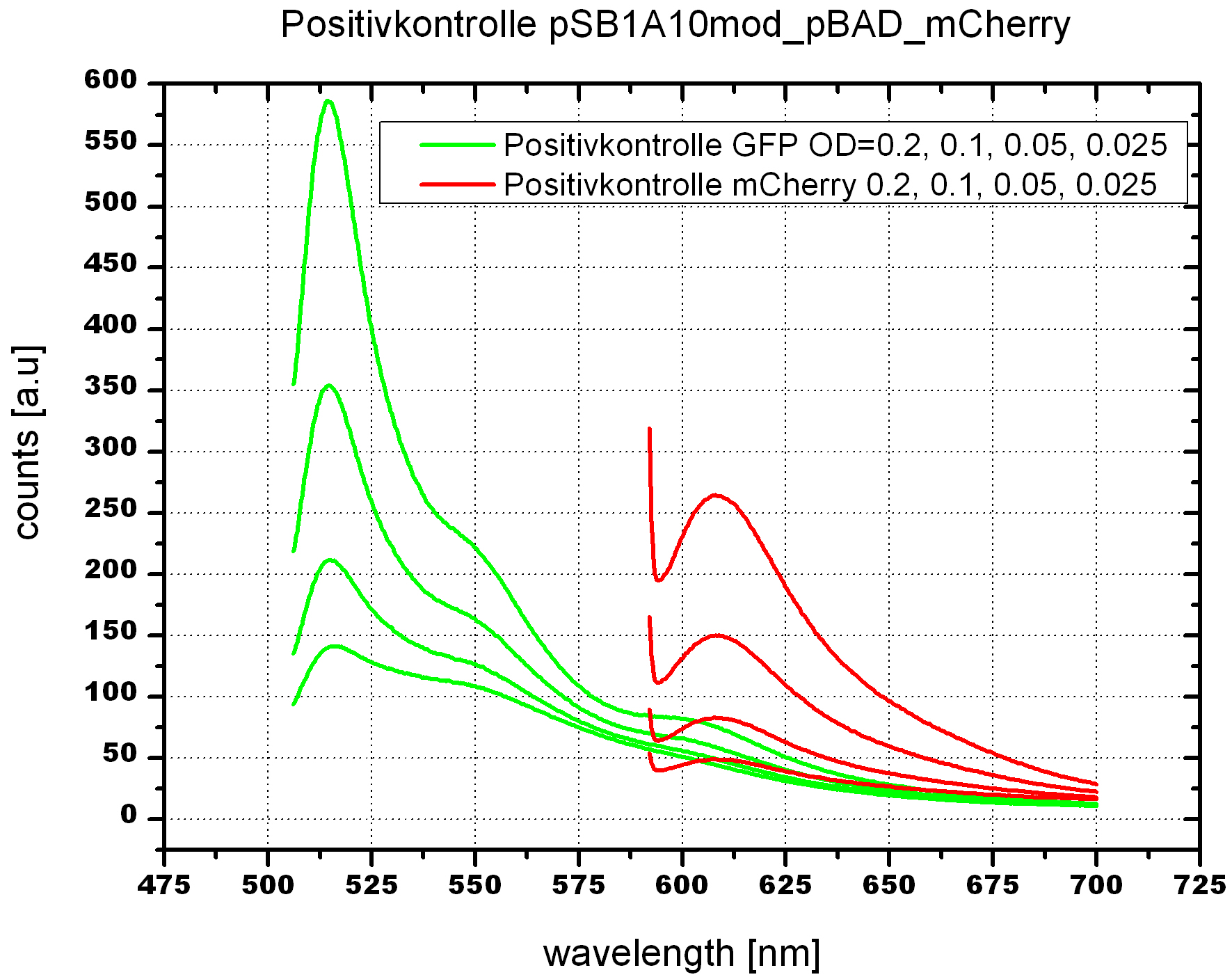
- 5ml cultures of BL21 cells
- pSB1A10mod_Pbad_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry
- pSB1A10mod_Pbad_HisTerm_mCherry_HisSig
- pSB1A10mod_Pbad_TrpTerm_mCherry_TrpSig
- ColonyPCR
- picked 4 Colonies per Neg.TrpControl from 02.09' ligation plates
- PCR using Program 'colonyPCR'. Elongation time modified to 1:20min
09.09.2010
- Fluoresence measurement
- using BL21 cells
- samples:
- Positive control (pSB1A10mod_pBAD_mCherry)
- Negative control (pSB1A10mod_pBAD_HisTerm_mCherry)
- pSB1A10mod_pBAD_HisTerm_mCherry_HisSignal
- pSB1A10mod_pBAD_TrpTerm_mCherry_TrpSignal
- Induction with 0.4% L-arabinose and 1mM IPTG
- Timepoints: 1 h, 2 h, 4 h, 10 h, 16 h (induced the day before)
- (OD checked seperately each time; average ODs=0.02-0.06)
- 30% glycerol stocks of BL21 cells carrying:
- Positive control (pSB1A10mod_pBAD_mCherry)
- Negative control (pSB1A10mod_pBAD_HisTerm_mCherry)
- pSB1A10mod_pBAD_HisTerm_mCherry_HisSignal
- pSB1A10mod_pBAD_TrpTerm_mCherry_TrpSignal
10.09.2010
- Fluorescence measurements
- Induction with 0.4% L-arabinose and 1 mM IPTG
- Measurement of OD600
- Fluorescence measurement at 15 h and 26 h
- Samples:
- pSB1A10mod_Pbad_mCherry - Positive Control
- pSB1A10mod_Pbad_HisTerm_mCherry - HisNeg. Control
- pSB1A10mod_Pbad_HisTerm_mCherry_HisSignal
- pSB1A10mod_Pbad_TrpTerm_mCherry_TrpSignal
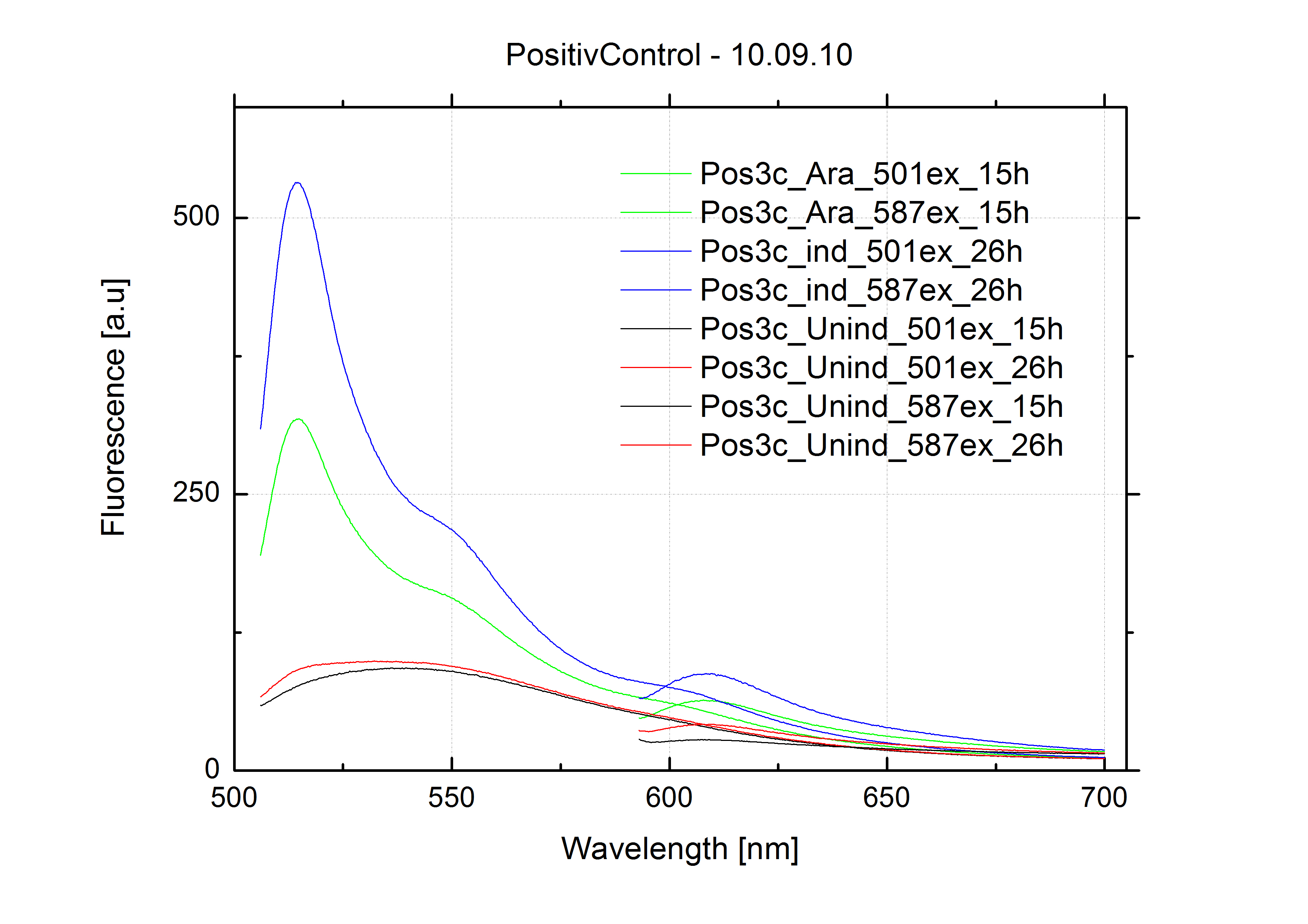
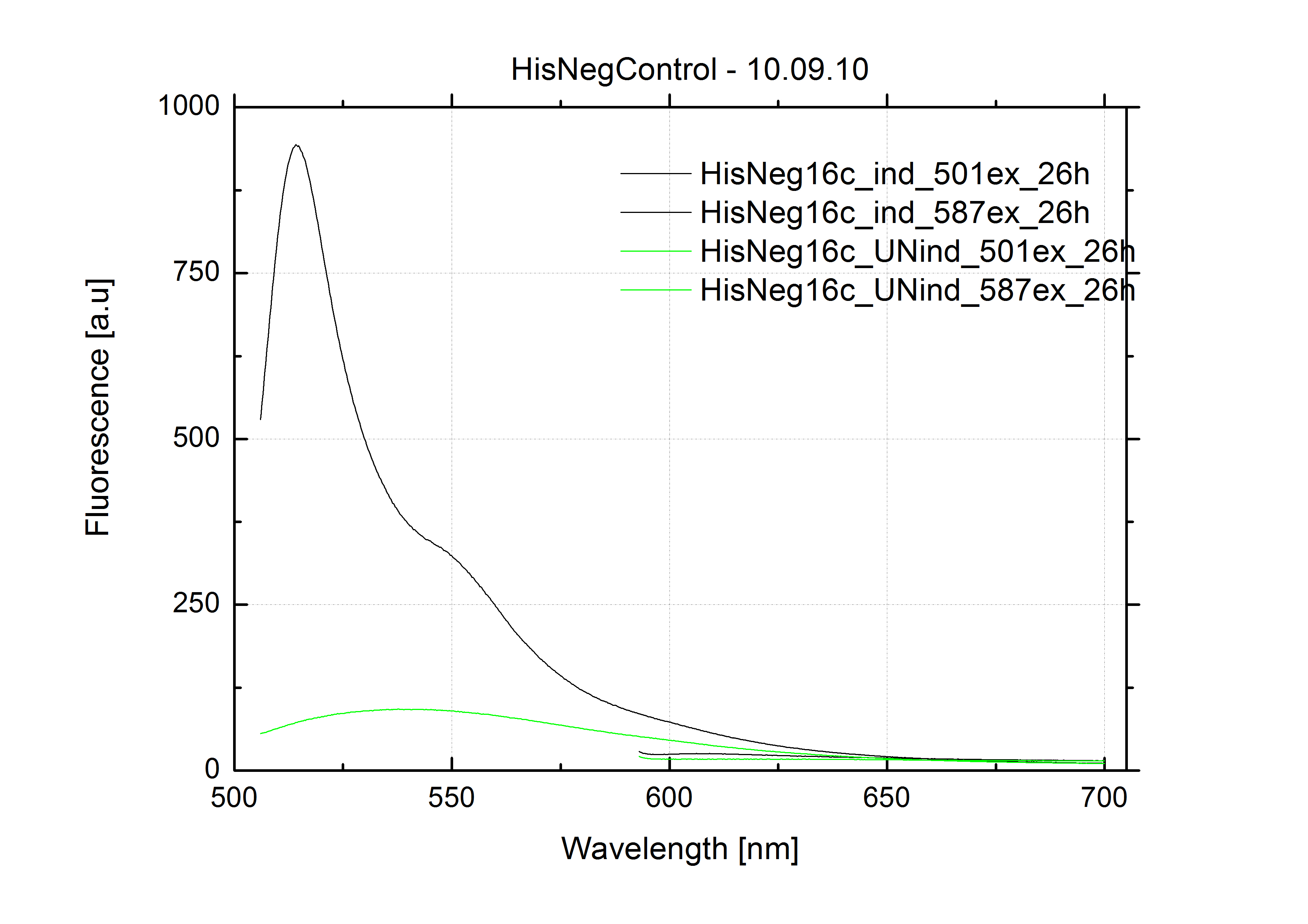
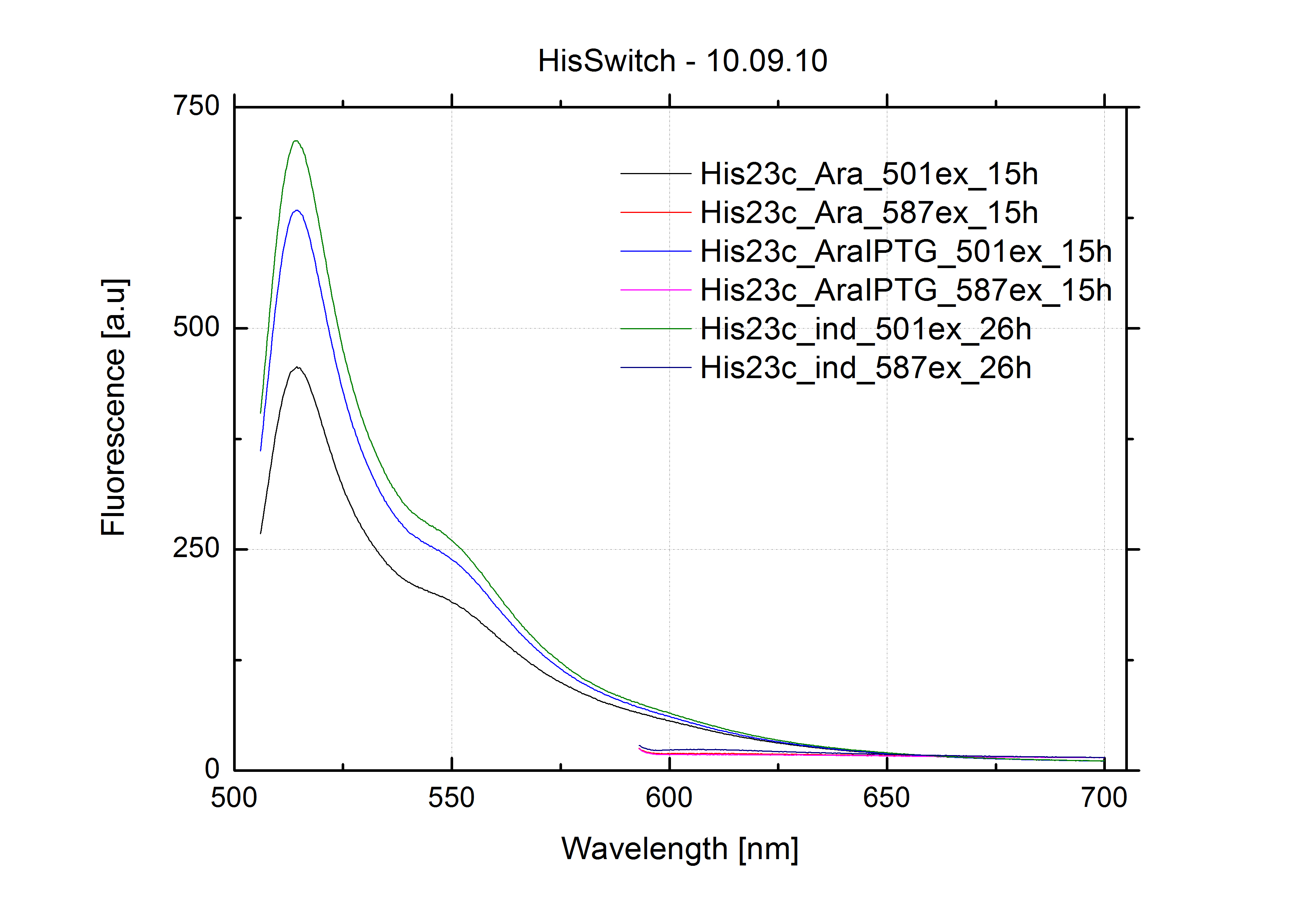

His-Switch DOES NOT seem to work!!! Measurements of Trp-Switch look weird, since Ara-induced culture showed strong mCherry signal than Ara+IPTG-induced culture. Maybe growing condition were not sufficient. We will use 50 ml flask next time!
Close
Week25
Cloning HisTerm/TrpTerm Ordering Aptamer New Measurement Plasmid
Week26
Cloning of Malachit green constructs HisTerm/TrpTerm
20.09.2010
- PCR Purification using Qiagen MinElute Kit
- Trp-Signal
- His-Signal
- Trp-Switch
- His-Switch
- Digestions 37°C, 2h
- Trp-Signal XbaI/PstI
- His-Signal XbaI/PstI
- Trp-Switch EcoRI/PstI
- His-Switch EcoRI/PstI
- --> Heat inactivation 20 min, 80°C
- Ligation
- pSB1A2_R0011 SpeI/PstI + Trp-Signal XbaI/PstI
- pSB1A2_R0011 SpeI/PstI + His-Signal XbaI/PstI
- pMalachit EcoRI/PstI + Trp-Switch EcoRI/PstI
- pMalachit EcoRI/PstI + His-Switch EcoRI/PstI
- pMalachit XbaI/SpeI
- Transformation of DH5a cells with ligation
21.09.2010
- Picked 2 colonies per plate of yesterday's transformation => we call them z-Series
- ColonyPCR of z-Series
- analytical agarose gel (2.5%)
- => ColonyPCR did not work. Probably not enough template.
- Picked futher colonies
- 2nd ColonyPCR of z-Series
22.09.2010
- analytical agarose gel (2.5%) of 2nd colonyPCR of z-Series
- pSB1A2-R0011-TrpSignal looks good. We removed all pSB1A2-R0011-HisSignal samples since there is a contamination on the gel and colonies turned redish. No conclusion regarding the other ligations of z_series can be drawn. LB control produces similar bands as to what we expected => Choose new Primers for PCR.
- 3rd colonyPCR of z-Series (samples 1-16z)
- used Primers Apt_For and Apt_Part_woT instead of BioBrick Primers (G1005 and G1004). Bands should be about 100bp longer.
- analytical agarose gel (2%)
23.09.2010
- PCR to obtain DNA for malachite green in vitro measurements
- 2z (XbaI/SpeI religated positive control)
- 16z (pMalachitApt_HisSig positive control)
- each samples was amplificated with two sets of primers (including and excluding the terminator BB0014):
- Apt_For and AptFull_wT_Rev
- Apt_For and AptPart_woT_Rev
- Purified with Qiagen MinElute Kit
- analytical digestion of z-series ligation:
- 2z (XbaI/SpeI religated positive control)
- 5z (pMalachitApt_TrpTerm)
- 12z (pMalachitApt_HisTerm)
- 16z (pMalachitApt_HisSig positive control)
- 28z (pSB1A2_R0011_TrpSig)
- NsiI only (HisTerm contains one cutting site for NsiI, so does pMalachiteApt => we expect 2 fragments):
- 12z (pMalachitApt_HisTerm)
- 10 µl ligations using digested samples from 21.09.2010
- pSB1A2_R0011 (SpeI/PstI cut) + HisSig (XbaI/PstI cut)
- pMalachiteApt (EcoRI/PstI cut) + TrpSwitch (EcoRI/PstI cut)
- pMalachiteApt (EcoRI/PstI cut) + HisSwitch (EcoRI/PstI cut)
- Transformed DH5a cell with 8µl of the above ligation samples
24.09.2010
- Fluorescence measurement: Malachite Green Assay
- 2z positive control (X/S religated: tac_MalachitApt) (PCR-amplified without BB0014 Terminator)
- 2z positive control (X/S religated: tac_MalachitApt_BB0014) (PCR-amplified with BB0014 Terminator)
- negative control (tac_BB1006Switch_MalachitApt_BB0014) (PCR-amplified with BB0014 Terminator)
- tac_BB1006Switch_MalachitApt + tac_BB1006Signal (both PCR-amplified without BB0014 Terminator)
- 2 µl sigma70 satured RNA E.Coli Polymerase (epicenter Biozyme) (2U)
- ca. 1µg DNA template
- 5 µM Malachite Green
- 10 µM DTT
- 4 µM NTPs
- 40 mM Tris-HCl pH = 7.1 @ 37°C; 7.5 @ 22°C
- 10 mM MgCl2
- 150 mM KCl
- added H20 to total volume of 100 µl
- Kinetics measurement @ 37°C
- Excitation 630 nm
- Emission 655 nm
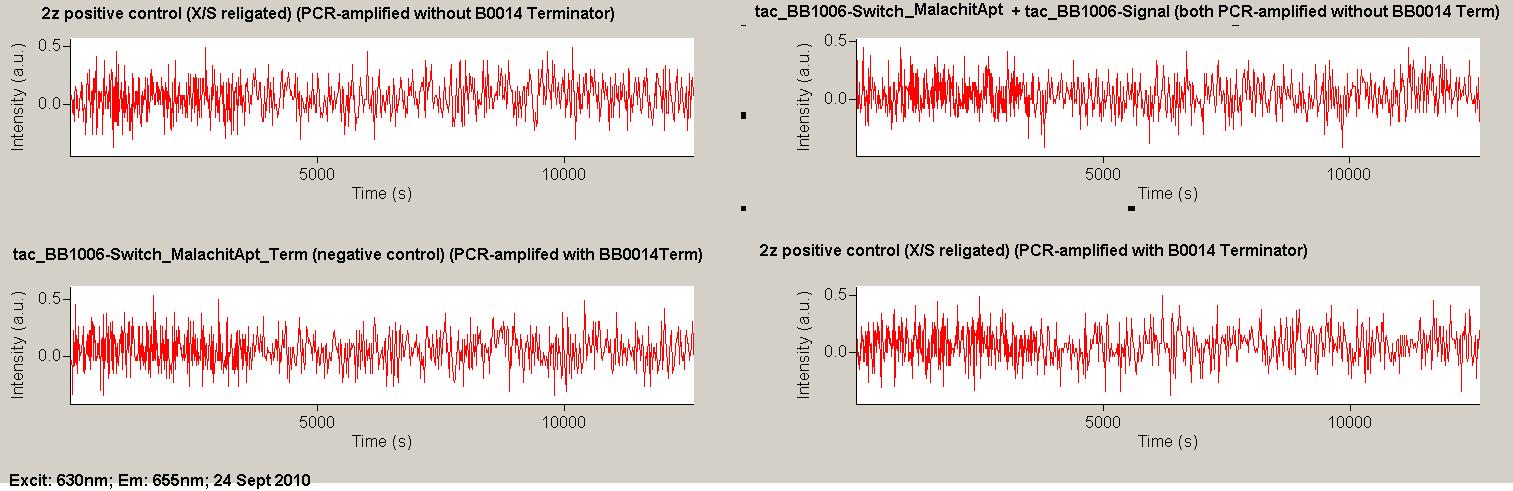
-
- no apatmer formation observed => positive controls did not work at all => assay did not work!!
-
- Exitation: 630 nm
- Results:
- no detactable peaks between 640nm and 800nm in any sample at any measured time (Settings were checked by detecting scattering peaks).
Close
Week27
T7 Switch
27.09.2010
- Fluoresence measurements
- Cary Spectrometer
- samples:
- with T7 RNA Polymerase (NEB)
- T7 positive control (T7Promotor_MalachitAptamer)
- with Epicenter E coli RNA Polymerase
- 2z positive control (X/S religated: tac_MalachitApt) (PCR-amplified without BB0014 Terminator)
- 2z positive control (X/S religated: tac_MalachitApt_BB0014) (PCR-amplified with BB0014 Terminator) (NO KINETICS RECORDED)
- with in vitro kit (cell lysate)
- 2z positive control (X/S religated: tac_MalachitApt) (PCR-amplified without BB0014 Terminator)
- 2z positive control (X/S religated: tac_MalachitApt_BB0014) (PCR-amplified with BB0014 Terminator)
- Scanning measurements: excitation at 630nm
- Kinetics measurements: excitation at 630nm; Emission at 655nm
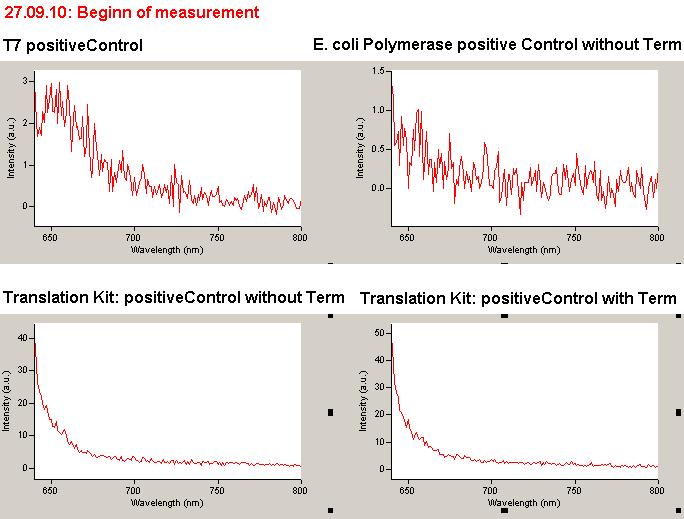
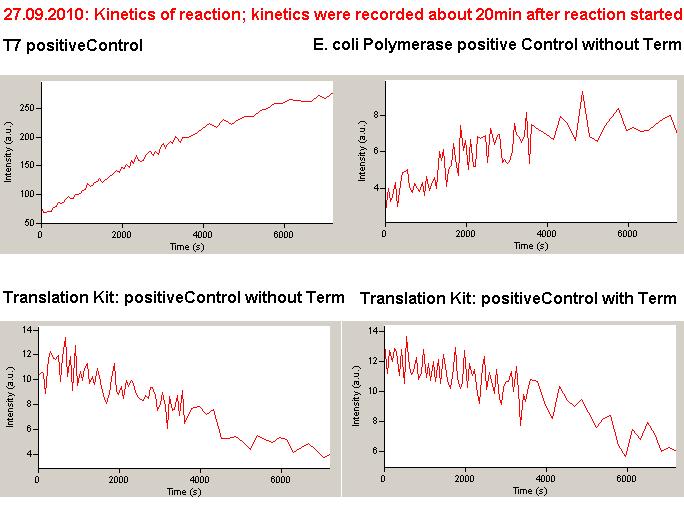
Results:
T7 Polymerase works perfectly. E coli Polymerase also produced RNA but much less (however enzyme might show reduced activity due to storage problems). In vitro (Cell lysate) kits do not work at all.
01.10.2010
T7-Measurments
- PCR of switch_phi_T7-construct to obtain dsDNA (conditions: igemPCR and 30s elongation time). Samples were purified using Qiagen MinElute.
- 1th measurment:
Maxi´s NEB buffer conditions: 40 mM Tris pH 7.4 @ RT, 40mM Mg2Cl, 5 µM Malachit-green, 4 mM NTPs, 2,5 U RNA-Polymerase
All DNA templates were all added to a final concentration of 200nM.
- sample 1: Positive control
- sample 2: negative control (= switch without any signal) (new DTT used for this sample)
- sample 3: switch + SigA1a
- sample 4: switch + SigA1c
- Results
- kinetic results
- Spectra
=> Switches do look quite good. However DTT was not the same in all samples.
- 2th measurment:
- "Paper´s" buffer conditions: 40 mM Tris pH 7.9 @ RT, 6mM Mg2Cl, 5 µM Malachit-green, 100 mM KCl, 0.8 mM NTPs, 2,5 U RNA-Polymerase
- sample 1: Positive control
- sample 2: negative control (= switch without signal)
- Maxi´s NEB buffer conditions: 40 mM Tris, 40 mM Mg2Cl, 10 µM Malachit-green, ph 7.4 @ RT
- sample 3: Positive control (new DTT used for this sample)
- sample 4: Negative control (= switch without signal)
Close
Week28
T7 Switch
04.10.2010
- 1st fluorescence measurement
-
- 200 nM T7-Switch (100 nM each strand) + SignalA_1a
- 200 nM T7-Switch (100 nM each strand) + SignalA_2a
- 200 nM T7-Switch (100 nM each strand) + SignalA_2b
- 200 nM T7-Switch (100 nM each strand) + SignalA_2c
- 40 mM Tris pH 7.1
- 40 mM MgCl2
- 10 mM DTT
- 1.6 mM NTPs
- 5 µM Malachite green
- 2.5 U T7 RNA Polymerase
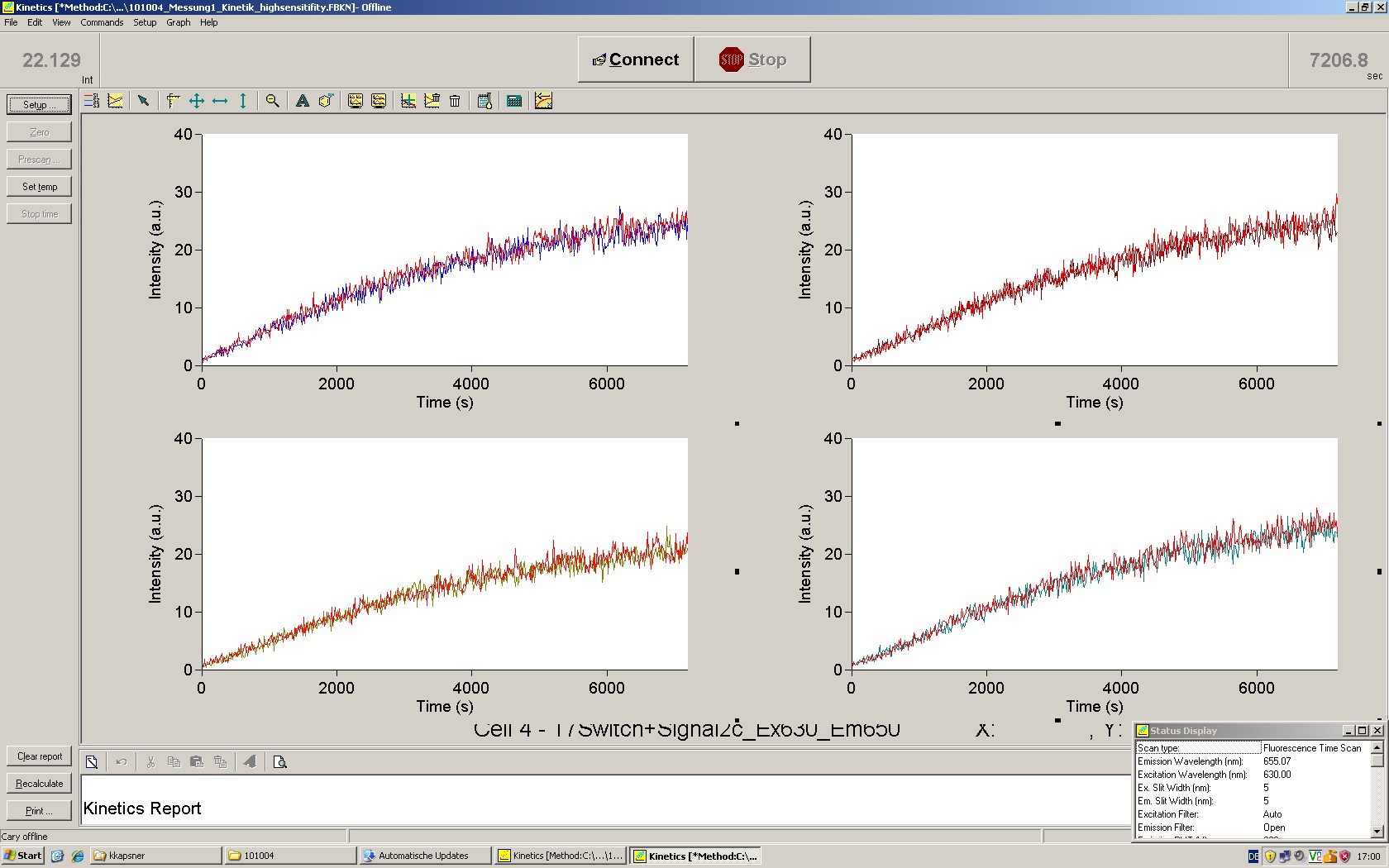
- Fluorescence increased in all samples in a very similar way, suggesting all signal to work equally well.
- 2nd fluorescence measurement
-
- 200 nM T7-Switch only (Buffer A)
- 200 nM T7-Switch only (Buffer B)
- positiv control (T7Promoter_MalchitAptamer) (Buffer A)
- positiv control (T7Promoter_MalchitAptamer) (Buffer B)
- 40 mM Tris pH 7.1 @ RT
- 40 mM MgCl2
- 10 mM DTT
- 1.6 mM NTPs
- 10 µM Malachite green
- 2.5 U T7 RNA Polymerase
- 40 mM Tris pH 7.9 @ RT
- 6 mM MgCl2
- 100 mM KCl
- 10 mM DTT
- 1.6 mM NTPs
- 10 µM Malachite green
- 2.5 U T7 RNA Polymerase
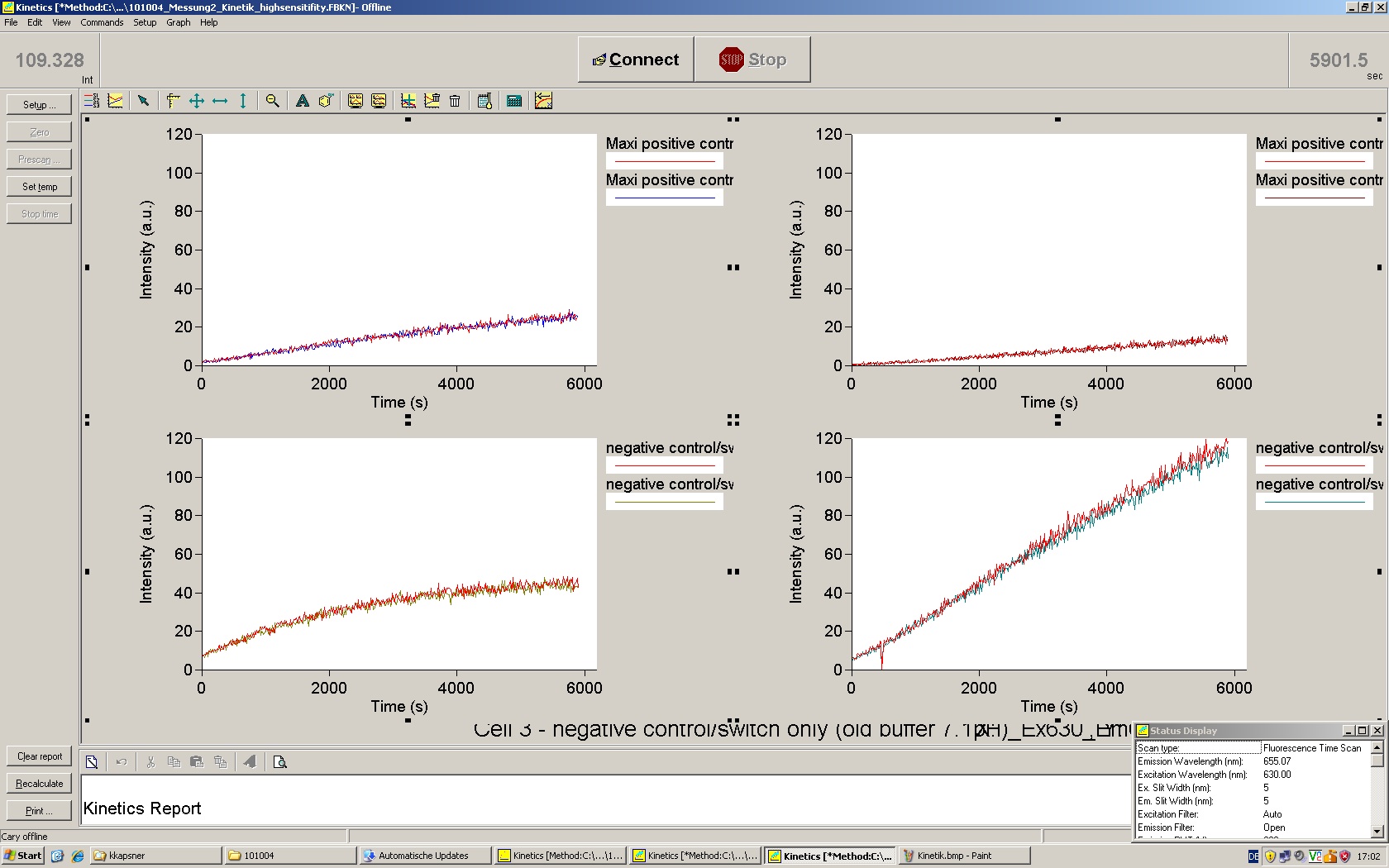
- Very strange results!! Both negative controls increased strongly compared to the positive controls that only slightly increased (buffer A seems to be better for transcription than buffer B).
- => Maybe something went very wrong. However, last Friday we observed a similar, strange behavior. Maybe the positive control is no good choice. It would make sence to use T7-Switch+SignalA_1a as a reference.
- Signals were generally stronger. 10 µM of malachite green seems to be quite good.
05.10.2010
- First measurement of the day: Signal, 1d, 2c, 3b
- Master Mix:
- 204 µl 2x Buffer
- 40.8 µl DTT
- 8.16 µl NTPs
- 21.05 µl Switch (1.94 µM)
- signals: # volume µl
- negative control - 1.213
- 1d - 1.290
- 2c - 1.450
- 3b - 1.376
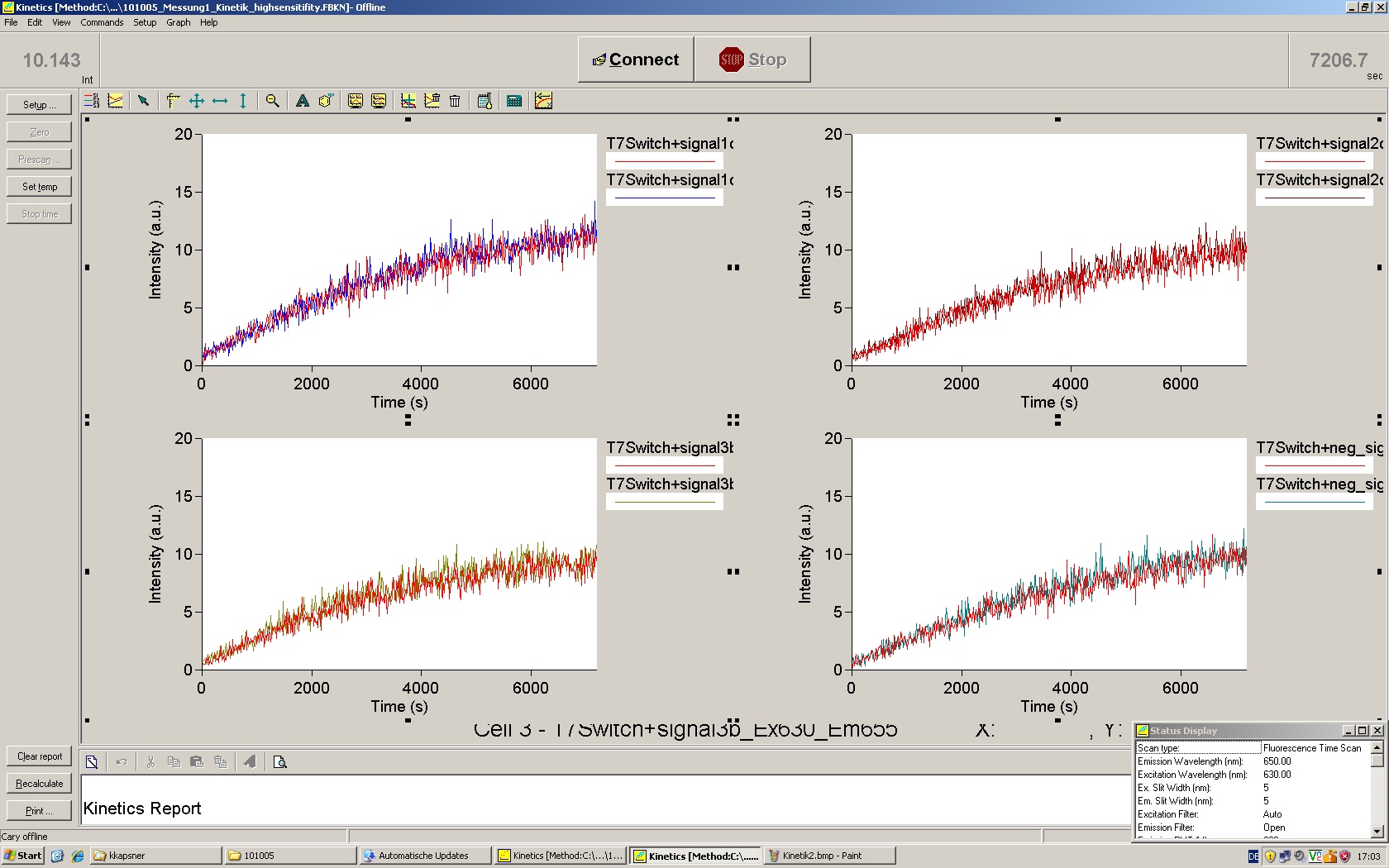
- PCR with positive control and T7 switch
PCR T7 switch/positive signal
- 32 x Mastermix: 1600 µl total volume
- 32 µl dNTPs
- 32 µl T7 forward primer
- 32 µl T7 reverser primer
- 160 µl 10x buffer
- 96 µl MgCl2
- 6.4 µl Taq Polymerase
- 10 µl Template (some older PCR)
- 1209.6 µl H2O
- Second measurement of the day: Signal negative control, nonsense signal, 1a (with 400 µM signal), 1a (with 2 µM signal)
- *** Master Mix:
- 204 µl 2x Buffer
- 40.8 µl DTT
- 8.16 µl NTPs
- 21.05 µl Switch (1.94 µM)
- signals: # volume µl
- negative control - 30.34
- 1d - 1.213
- 2c - 2.672
- 3b - 13.36
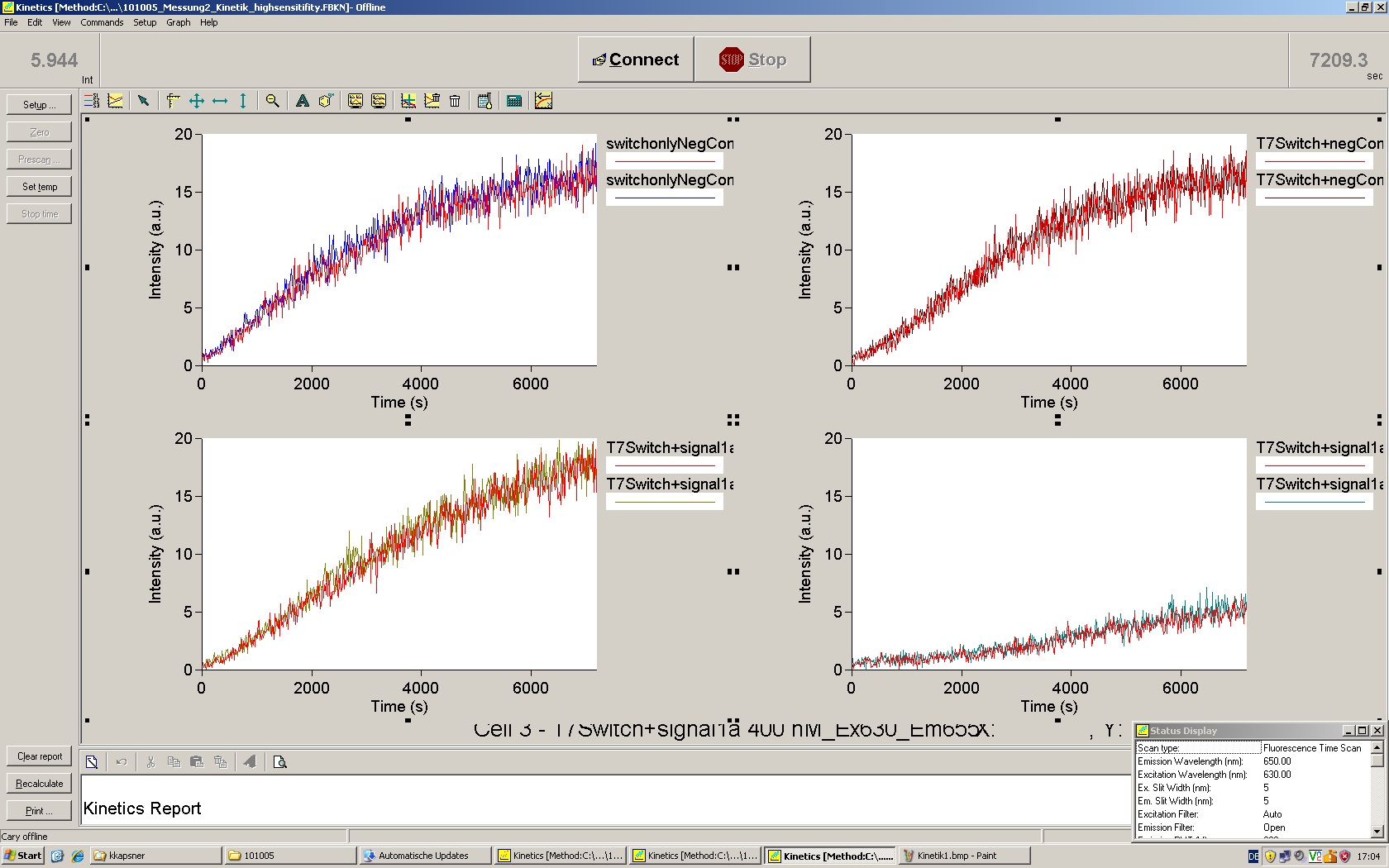
- Evaluation of different malachite green concentrations
- 5 µM, 10 µM, 15 µM, 25 µM chosen
- high temperature dependency
- Equilibration of samples important
06.10.2010
- PCR
- yesterday's PCR Purification using Zymo Clear and Concentrated
- yields
- Switch: 164 ng/µl
- positive control: 28 ng/µl
- problems with the temperature? Yield too low!
- PCR of positive control
- annealing temperature set to 48°C
- 20 x Mastermix:
- 20 µl dNTPs
- 20 µl T7 forward primer
- 20 µl T7 reverser primer
- 100 µl 10x buffer
- 60 µl MgCl2
- 4 µl Taq Polymerase
- 1 µl Template (some older PCR)
- 775 µl H2O
07.10.2010
- T7 Trancription
- new buffer:
- Stocks, volume for 20 ml, endconcentration in 1x
- 1M Tris/HCl, 1.6 ml, 40 mM
- 500 mM MgCl2, 3.2 ml, 40 mM
- 250 mM malachite green, 1.6 ml, 20 µM
- H20
- Measurement
- 4.1 x, switch, switch + nonsense, switch + 1a, switch + 1c
- 21.16 µl switch, 1.93 µM
- 10.25 µl RPO
- 205 µl buffer
- 41 µl DTT 100 µM
- 20.5 µl rNTPs
- H2O
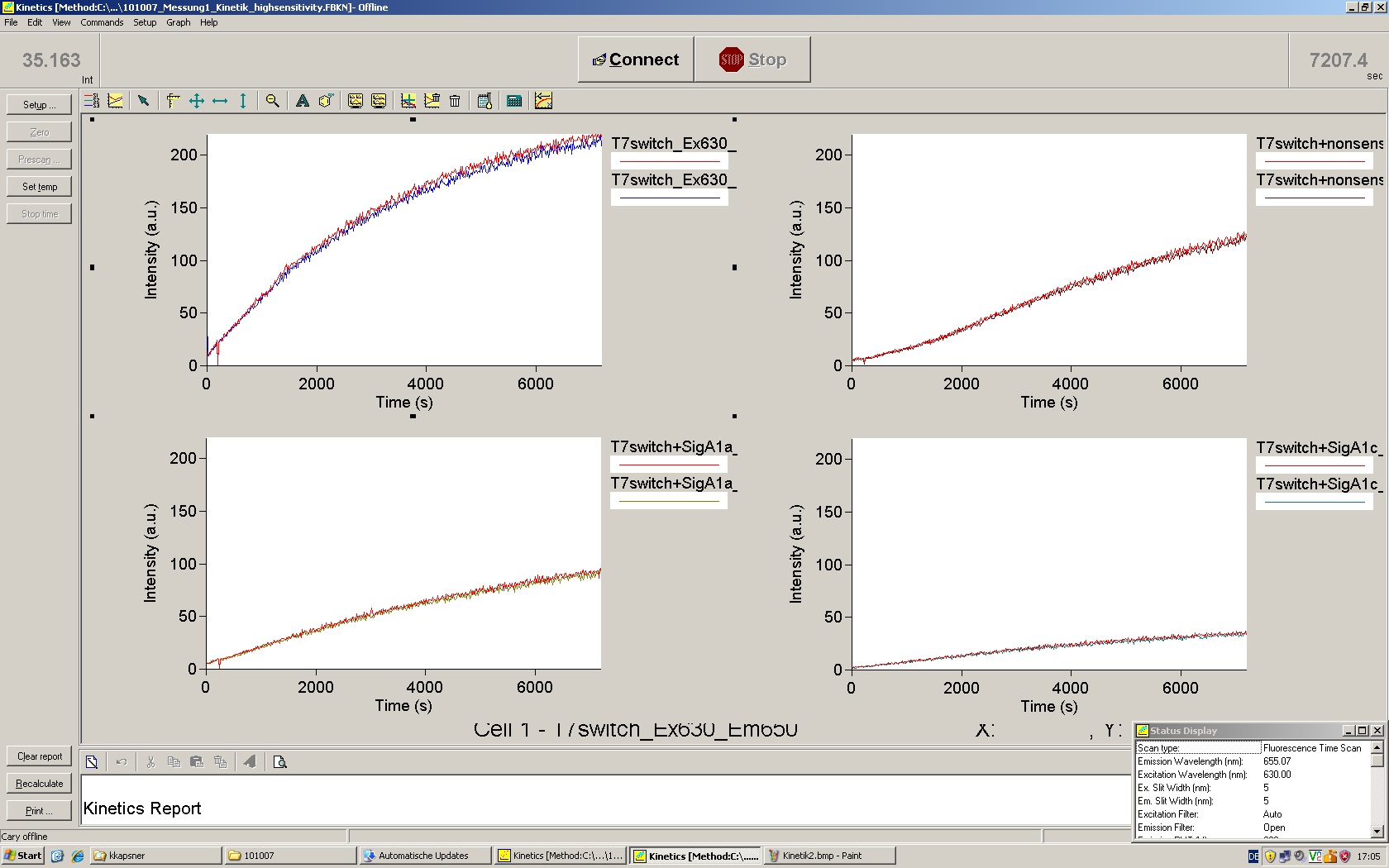
- second measurement
- Measurement
- 4.1 x, switch, switch + nonsense, positive control + 1b, positive control + 1b
- 21.16 µl switch, 1.93 µM
- 10.25 µl RPO
- 205 µl buffer
- 41 µl DTT 100 µM
- 20.5 µl rNTPs
- H2O
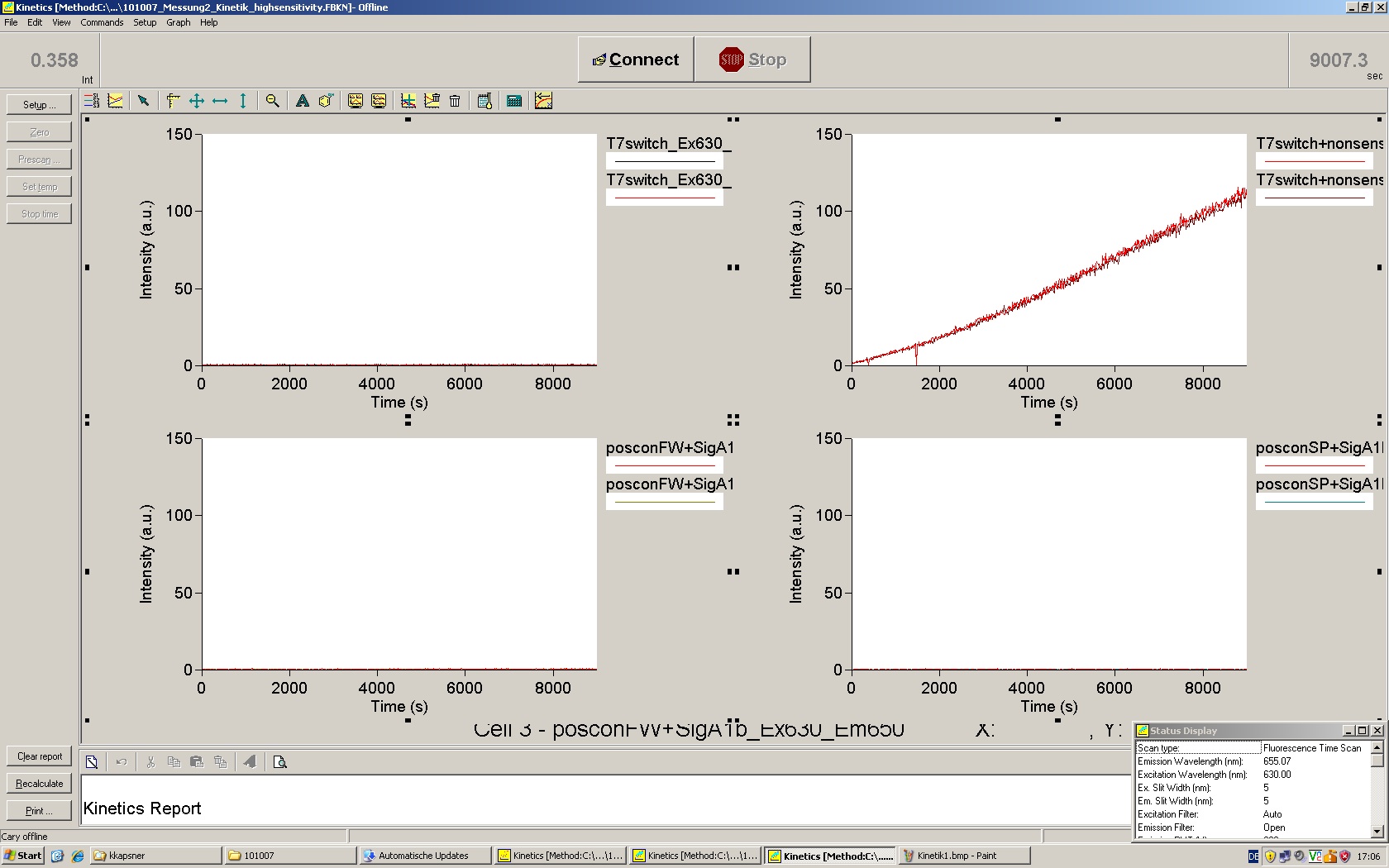
08.10.2010
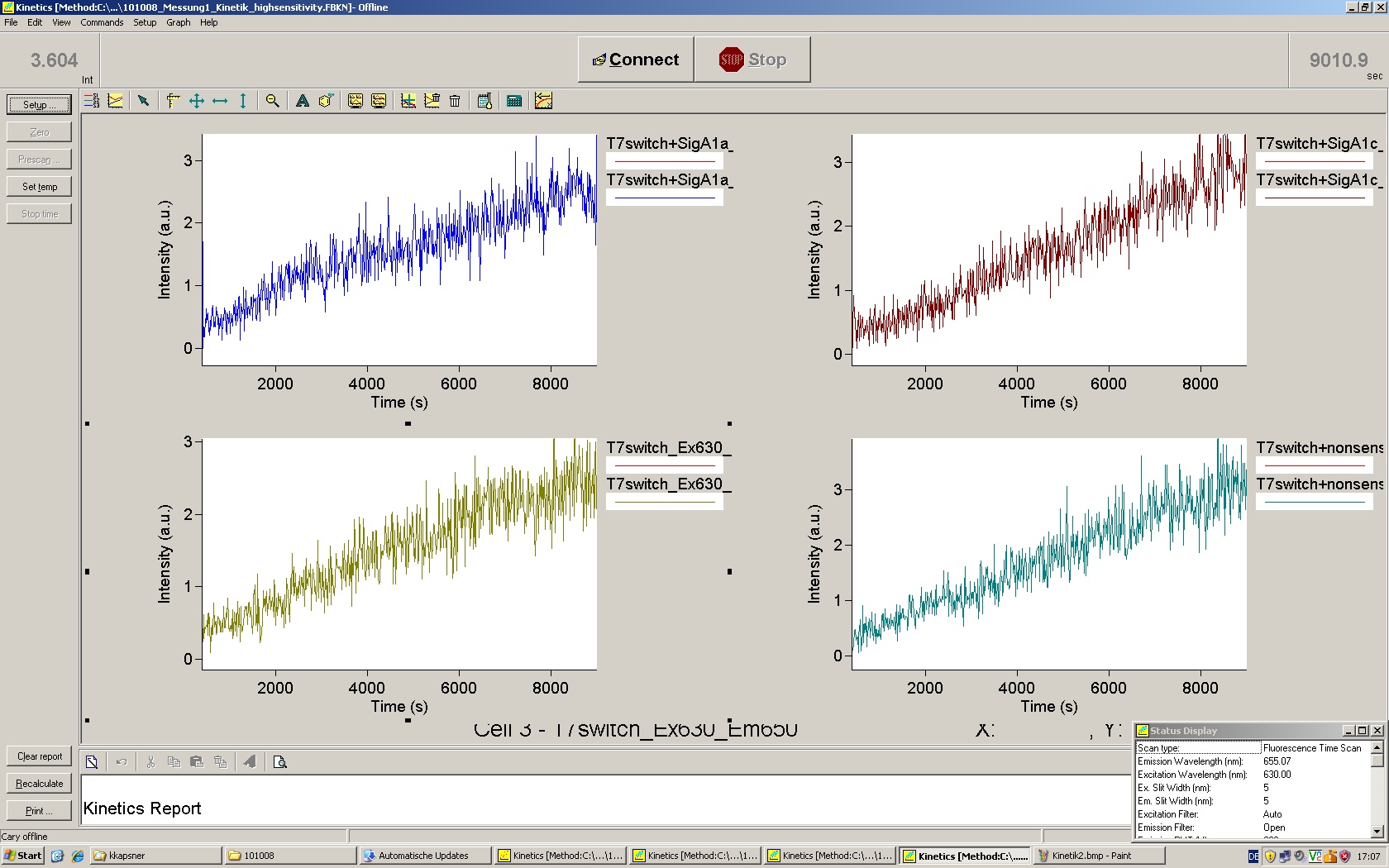
Close
Week29
T7 Switch
11.10.2010
- PCR T7 switch
- 32 x Mastermix: 1600 µl total volume
- 32 µl dNTPs
- 32 µl T7 forward primer
- 32 µl T7 reverser primer
- 160 µl 10x buffer
- 96 µl MgCl2
- 6.4 µl Taq Polymerase
- 10 µl Template (some older PCR)
- 1209.6 µl H2O
- Standart iGEM PCR program:
- 95°C, 2'
- 1) 95°C, 0,5'
- 2) 58°C, 0.5'
- 3) 71°C, 1'
- 71°C, 7'
- repeat 1)-3) 35 times
- yield after purification: 80 µl, 1.14 µM
- Malchitegreen measurement
- for 4.1 x 100 µl
- 35.88 µl PCR product
- 20.05 dNTPs
- 41 µl DTT
- 10.25 µl T7 RPO
- 8.75 µl switch
- 2 µl signal per 100 µl
- measured signals: (1) nonsense, (2) 1a, (3) 1b, (4) 2a
- Voltage set to 1000 (maximum)
- 38°C, every 15 s measurement
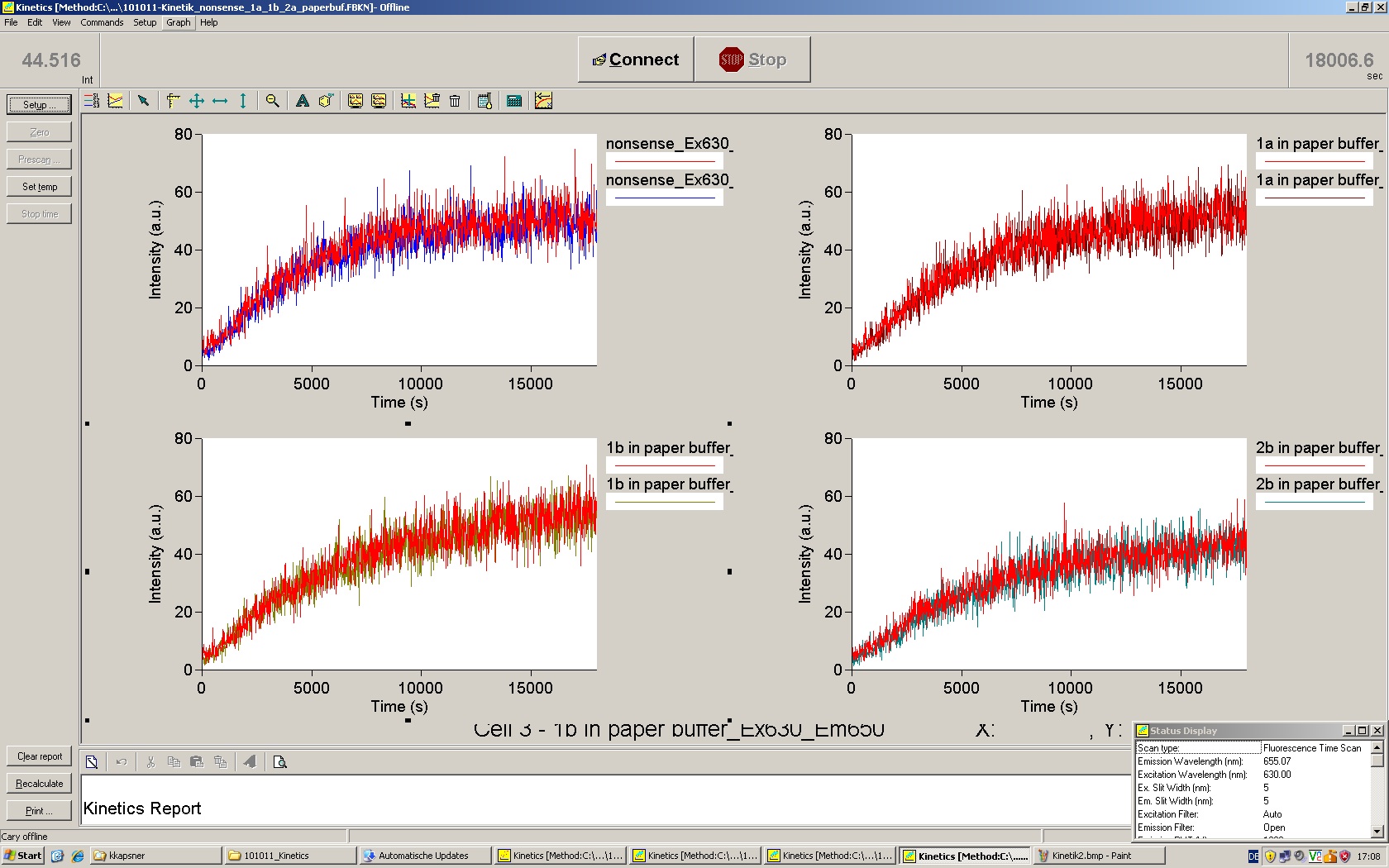
- start at 5 au, end at 25 au
- rise visible over time in all samples, nonsense signal with the fastest rise, 1a similiar, end signal similiar in all measurements
12.10.2010
- PCR T7 switch
- 32 x Mastermix: 1600 µl total volume
- 32 µl dNTPs
- 32 µl T7 forward primer
- 32 µl T7 reverser primer
- 160 µl 10x buffer
- 96 µl MgCl2
- 6.4 µl Taq Polymerase
- 10 µl Template (some older PCR)
- 1209.6 µl H2O
- changed iGEM PCR program:
- 95°C, 2'
- 1) 95°C, 0,5'
- 2) 50°C, 0.5'
- 3) 71°C, 1'
- 71°C, 10'
- repeat 1) - 3) 35 times
- --> melting temperature primers: 55/54°C!!!
- yield after purification: 300 ng/µl in 60 µl, 3.66 µM
- Malachitegreen measurement with preincubation of transcription stuff with signals
- for 4.1 x 100 µl
- 35.88 µl PCR product
- 20.05 dNTPs
- 41 µl DTT
- 10.25 µl T7 RPO
- 2 µl signal per 100 µl
- preincubation of signal with RPO for one hour, 37°C
- addition of signal after one hour (2.735 µl)
- once done in eppis (low bind), once in cuvettes
- no rise in both
- incubation and measurement of cuvette-incubated mix over night: no rise visible
- 15 % denaturing acrylamide gels
- for 50 ml
- 15 % acrylamide: 18.75 ml 40 % acrylamide
- 6M urea: 18 g
- 1x TBE: 5 ml 10x TBE
- 500 µl APS
- 50 µl TEMED
- H20 till 50 ml
- only 30 ml needed for two gels
- big combs for a lot of sample :)
- glass plates cleaned with RNAseZip before pouring the gel
13.10.2010
- in vitro T7 transcription, check by polyacrylamide gel electrophoresis
- 10 ml 5 x paper buffer
- 200 mM Tris/HCl, pH=7.85: 2 ml 1M Tris/HCl, pH=7.85
- 30 mM MgCl2: 600 µl 0.5 M MgCl2
- 500 mM KCl: 5 ml 1 M KCl
- 2.4 ml H2O
- for in vitro transcription
- 0.5 µl T7 RPO: 25 U
- 0.5 µl rNTPs : 20 mM
- 1.36 µl switch: 250 µM
- 1 µl signal: 250 µM
- 2 µl DTT: 10 µM
- 0.2 µl RNase inhibitor
- 4 µl 5x buffer
- 10.44 µl H2O
- 5.1 times
- no signal, nonsense, 1a, 1c, 2a
- in low bind tubes
- 2 hour, 37°C
- actually 2 hours and half a hour pocket cleaning time
- for PAGE:
- don't try to run yourgel in the pouring device - if you do so: feel very stupid and embarrassed (I guess I've ran over 100 gels in my life yet... still too stupid...)
- don't feel tempted to use the Dietz' group's 0.5 x TBE buffer - if you do so: feel very stupid and embarassed, discard buffer carefully and use 1 x TBE
- don't use a comb which is thinner than your spacers - if you do so: scrap gel pieces out of the pockets for half an hour
- cook your sample for 5 minutes, 95°C - if you do not so, feel stupid, embarassed and hope that it won't matter so much
- I did not cool the samples
- gel runs at 100 V (~ 10V/cm)
- 20 µl sample + 20 µl ambion loading buffer (1-2 x loading buffer - who does that?) --> 40 µl fits nicely
- 2 µl low molecular weight marker + 20 µl loading buffer + 18 µ H2O
- in 1 x TBE
- M, control (same amounts switch and signals as in the samples), no signal, random, 1a, 2a, 1c, random overnight, 1a overnight, 1c overnight
- overnight samples: measurement with malachitegreen sample over night: no rise visible
- Xylene Cyanol: Comigrating with 60, Bromphenol BLue: comigrating with 15 (http://www.protocol-online.org/cgi-bin/prot/view_cache.cgi?ID=845) - Maxi: bei Hälfte ungefähr
- run for 1:45 hours, Bromphenoleblue at about half of the gel
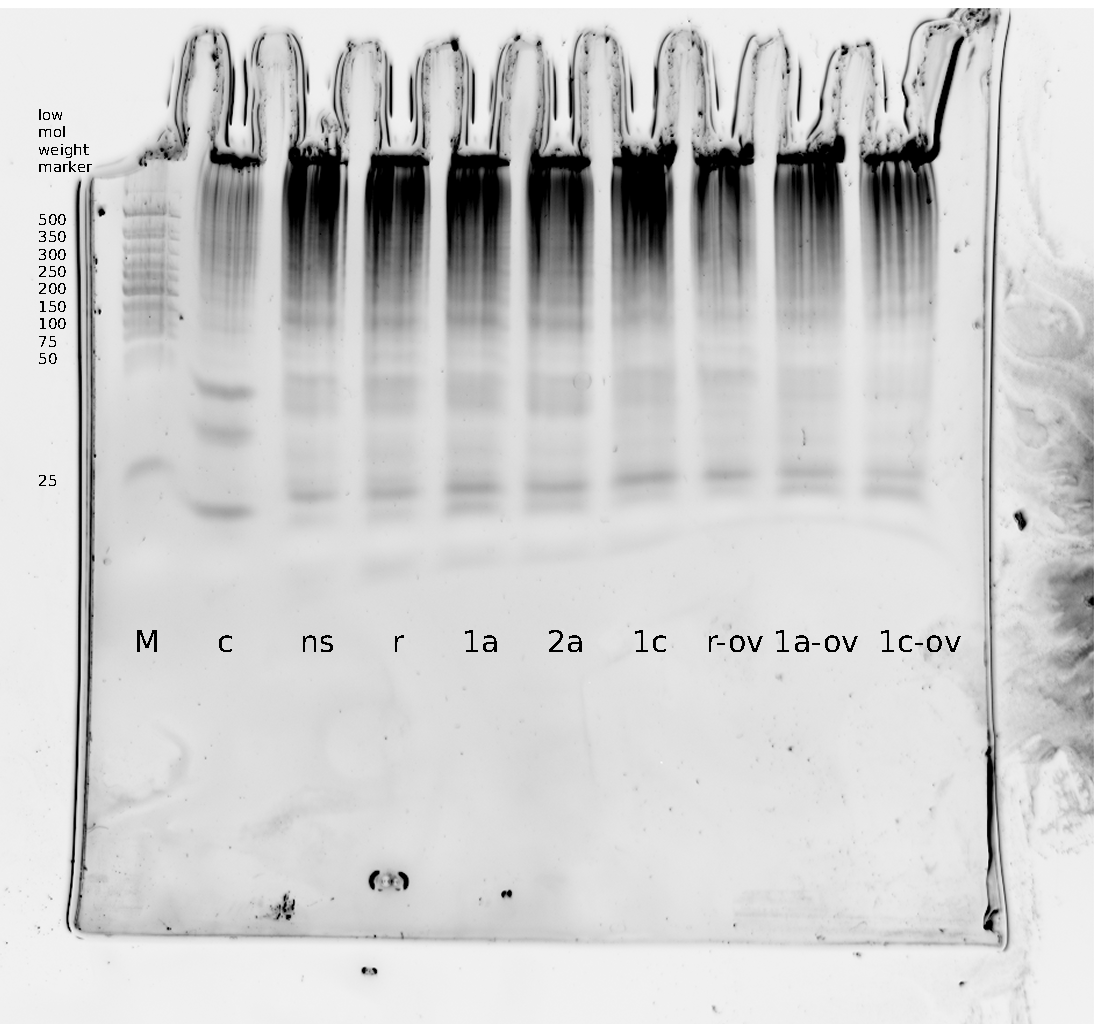
- --> c=control, ns=no signal, r=random
- --> weird smear everywhere: degraded RNA?
- --> no switch visible on the gel: 133 bp!
- --> signal length: 29-43 bp visible
- --> 25 bp: signal sense, between 50-25 bp: signals: 1a and 1c with approximately the same length, 2a is longer
- --> Wie Sie sehen, sehen Sie nichts.
- PCR of switch: Biomers original used as template
- PCR T7 switch
- 32 x Mastermix: 1600 µl total volume
- 32 µl dNTPs
- 32 µl T7 forward primer
- 32 µl T7 reverser primer
- 160 µl 10x buffer
- 96 µl MgCl2
- 6.4 µl Taq Polymerase
- 5 µl Template (some older PCR)
- 1214.6 µl H2O
- changed iGEM PCR program:
- 95°C, 2'
- 1) 95°C, 0,5'
- 2) 50°C, 0.5'
- 3) 71°C, 1'
- 71°C, 10'
- repeat 1) - 3) 35 times
14.10.2010
- yesterday's measurement
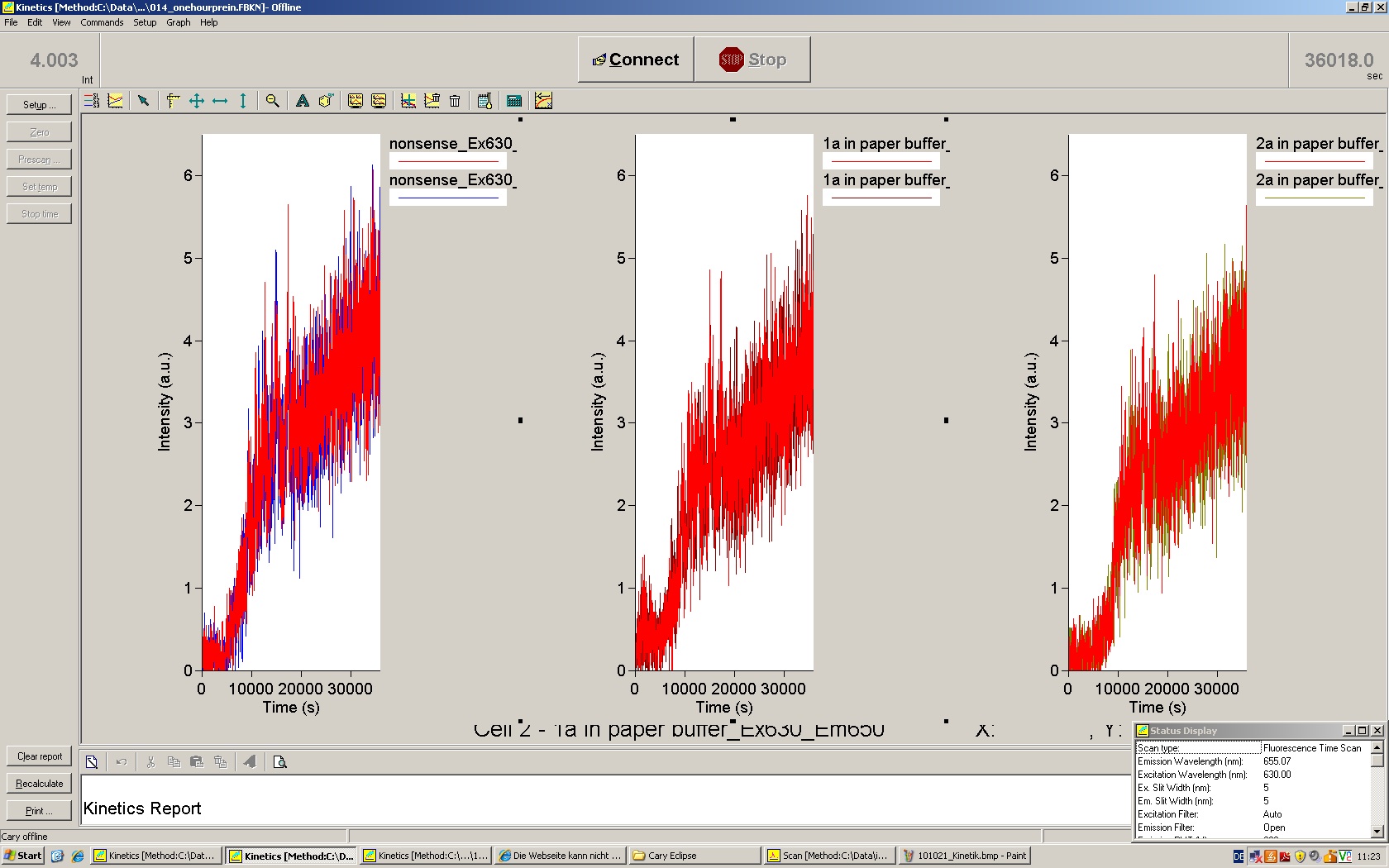
- rise visible, comparable to tuesday measurement
- samples frozen, put on 15 % PAGE
- purification of yesterday's PCR
- in vitro T7 transcription, check by polyacrylamide gel electrophoresis
- 10 ml 5 x paper buffer
- 200 mM Tris/HCl, pH=7.85: 2 ml 1M Tris/HCl, pH=7.85
- 30 mM MgCl2: 600 µl 0.5 M MgCl2
- 500 mM KCl: 5 ml 1 M KCl
- 2.4 ml H2O
- for in vitro transcription
- 0.5 µl T7 RPO: 25 U
- 0.5 µl rNTPs : 20 mM
- 1.36 µl switch: 250 µM
- 1 µl signal: 250 µM
- 2 µl DTT: 10 µM
- 0.2 µl RNase inhibitor
- 4 µl 5x buffer
- 10.44 µl H2O
- 5.1 times
- no signal, nonsense, 1a, 1c, 2a
- in low bind tubes
- 2 hour, 37°C
- actually 2 hours and half a hour pocket cleaning time
- 2 % agarose gel to check previous PCR products
- --> yesterdays results bad because no switch :)
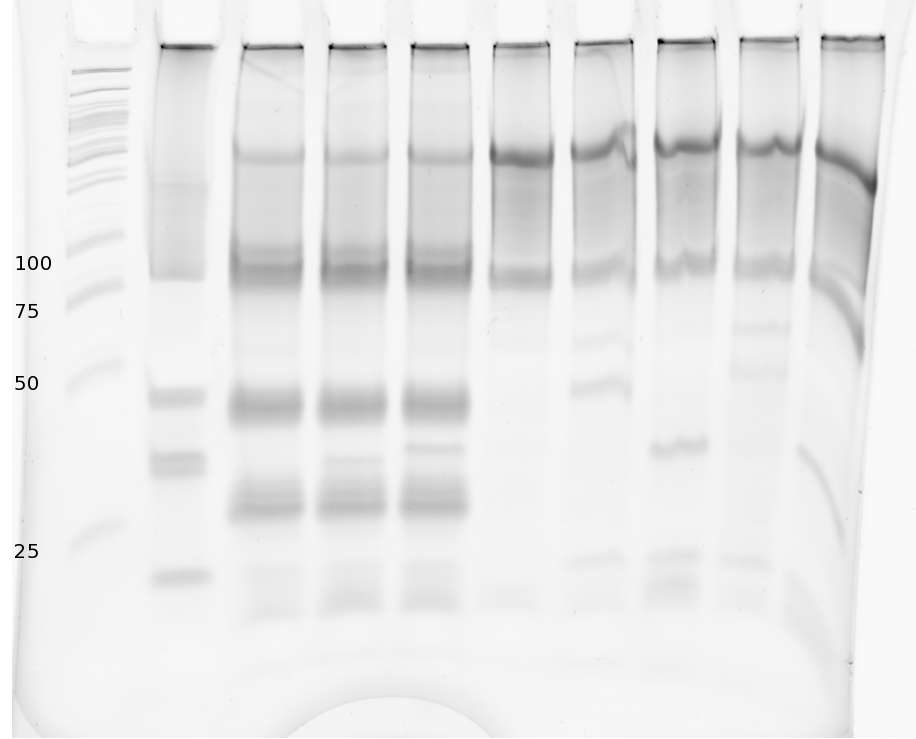
- --> M = low molecular weight marker (NEB)
- --> c=control=all DNAs mixed together in used concentrations: switch and all signals, ns=no signal, r=random=nonsense
- --> r, 1a, 2a on the left: overnight incubation with malchitegreen
- --> r, 1a, 1c, 2a on the right: two hour in vitro transcription without malachitegreen
- --> switch: 133 bp!
- --> signal length: 29-43 bp
- --> T7 promoter sense: ca. 20 bp (I can't look it up right now)
- --> extra bands visible after overnight transcription (rise in malchitegreen fluorescence visible)
- --> switch runs at a strange height: WHY? looks normal on 2 % agarose gel
- --> upper band: RPO bound to DNA? Compare EMSA
- --> DNA ladder, RNA bands: How to compare?
- Okay, let's try to interpret this:
- Denaturing conditions, everything precooked: No guarantee for double stranded DNA/RNA
- control: switch and signals from tubes on gel - otherwise treated the same
- --> internal standart: switch at about 80 bp (low molecular weight standart) equals 133 bp
- --> band seen in all samples at the height of random/1c --> termination product of switch??? (expected size: about 90 bp? does not fit at all?!)
- --> lower band visible, what is it?
- --> What is left: NO Differences Between Random control (=nonsense) and Designed Switches...
- New malachitegreen assay (overnight in vitro transcription) with new PCR product
- for 4.1 x 100 µl
- 35.88 µl PCR product
- 20.05 dNTPs
- 41 µl DTT
- 10.25 µl T7 RPO
- 2 µl signal per 100 µl
- preincubation of signal with RPO at 37°C
- no rise during preincubation (makes sense)
- addition of signal after about one and a half hour
15.10.2010
- in vitro transcription - malachite green
- slight rise visible after overnight incubation after preincubation of signal
- only 1/4 of the intensity measured yesterday
- spectra fit but very weak signal, scattered spectra
- Next step: addition of DNaseI (RNase free) to overnight transcription products
17.10.2010
- Malachite-green measuring assay
- 1x:
- 2 µl signal, 5 µM
- 4.66 µl switch, 176 µM
- 2.5 µl RPO
- 50 µl Paperbuffer
- 10 µl DTT 100 µM
- 5 µl rNTPs
- 25.84 µl H2O
- 4.1 x
- 19.11 µl switch, 176 µM
- 10.25 µl RPO
- 205 µl Paperbuffer
- 41 µl DTT 100 µM
- 20.5 µl rNTPs
- 105.94 µl H2O
- Signals: random, 1a, 2a, 1c
Close
Week30
Cloning of Parts into pSB1C3 T7 & E. coli
18.10.2010
- Yesterday's malachitegreen-binding assay
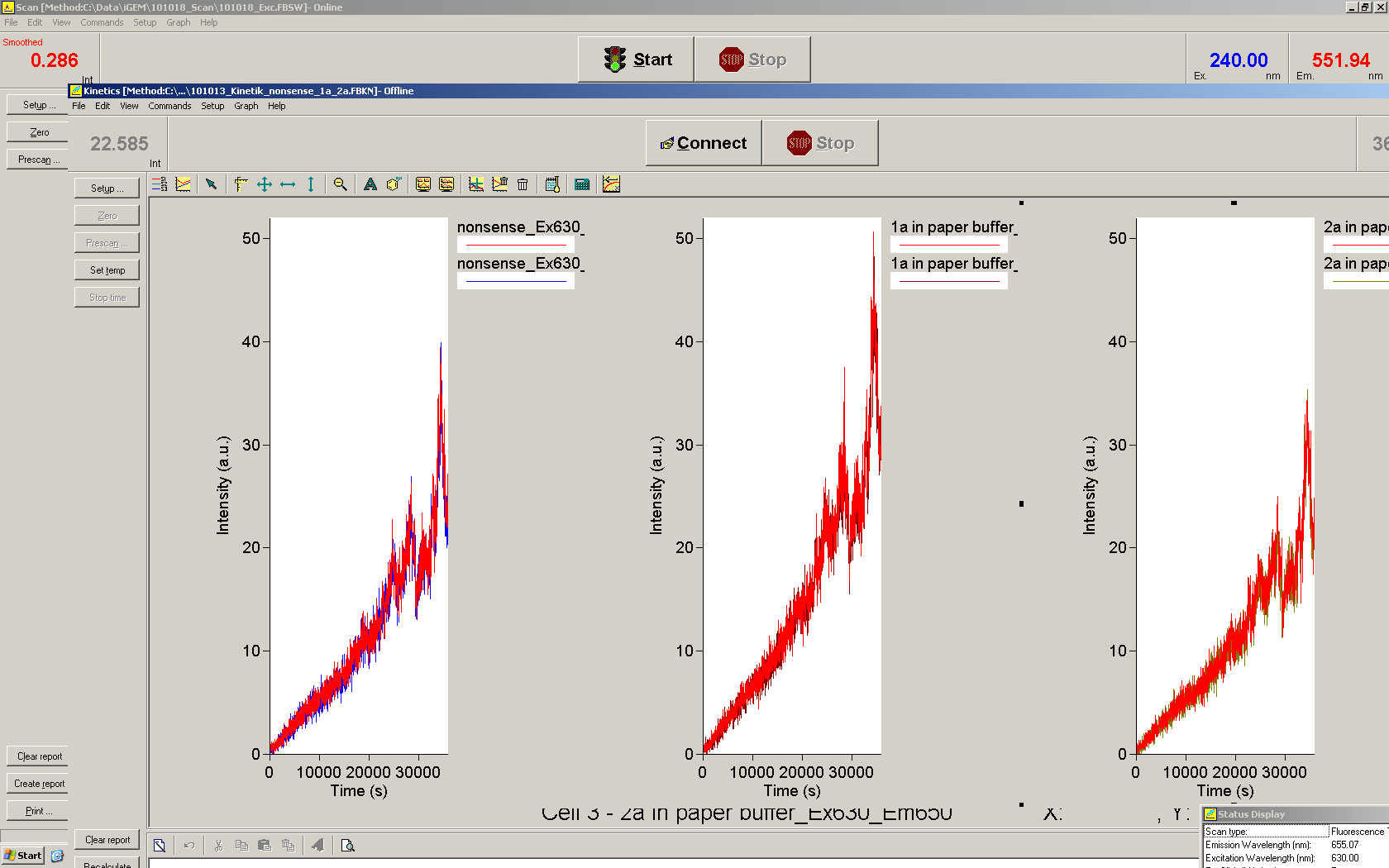
--> Favourite one ever so far!
- Emission and Excitation: Exc=530 nm, Em=552 nm
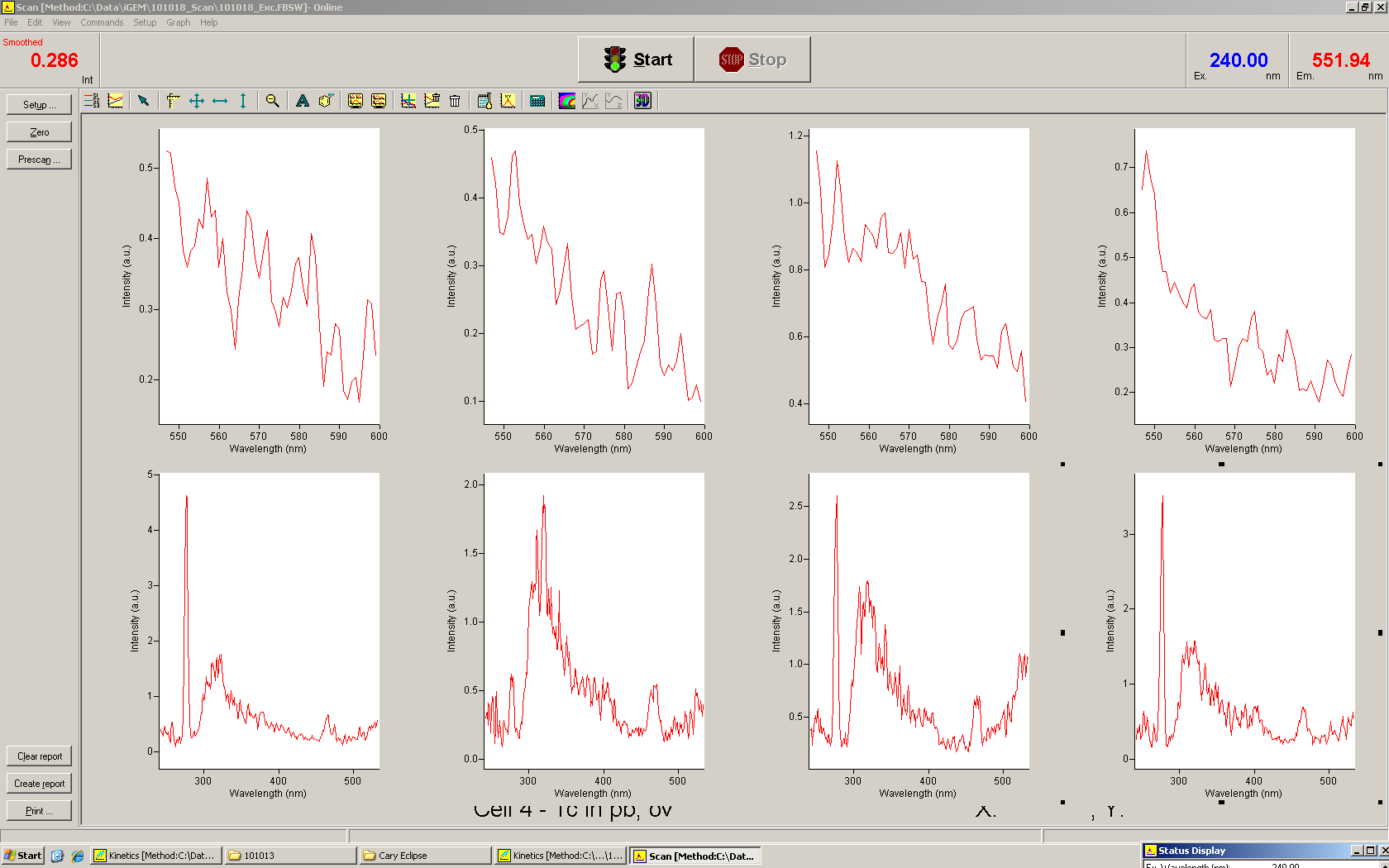
- Biomers order arrived
- DNA resolved according to manual, 100 µM endconcentration
- PCR T7 switch/positive signal
- 32 x Mastermix: 1600 µl total volume
- 32 µl dNTPs
- 32 µl T7 forward primer
- 32 µl T7 reverser primer
- 160 µl 10x buffer
- 96 µl MgCl2
- 6.4 µl Taq Polymerase
- 10 µl Template (some older PCR)
- 1209.6 µl H2O
- changed iGEM PCR program:
- 95°C, 2'
- 1) 95°C, 0,5'
- 2) 50°C, 0.5'
- 3) 71°C, 1'
- 71°C, 10'
- repeat 1) - 3) 35 times
- yields positive control:
- Mr=67234,6 g/mol
- 230 ng/µl --> 3.42 µM
- 198 ng/µl --> 2.94 µM
- 209 ng/µl --> used for cloning
- 178 ng/µl --> 2.65 µM
- yields switch:
- Mr=82059 g/mol
- 170 ng/µl --> 2.07 µM
- 204 ng/µl --> 2.49 µM
- 179 ng/µl --> 2.18 µM
- 160 ng/µl --> 1.95 µM
- 6 M urea 15 % acrylamid PAGE
- DNase I digestion
- 1 µl 10 x DNase buffer
- 1 µl DNaseI
- 20 µl reaction product from malachitegreen assay, 17.10.10
- 37°C, 90 minutes
- Gel:
- 20 µl loading buffer and 20 µl sample
-
- Malachitegreen assay with double stranded signals
- Concentration verification of sense and antisense in ng/µl
- diluted 1:8 (5+35 µl)
- signal - sense ng/µl - antisense ng/µl
- 1a - 1391 - 1584
- 2b - 1812 - 1823
- 1c - 2208 - 1575
- random2=nonsense2 - 2480 - 1903
- Concentration verification of sense and antisense in µM
- signal - sense µM - antisense µM
- 1a - 15.47 - 17.99
- 2b - 15.43 - 15.70
- 1c - 16.95 - 12.31
- r2 - 21.27 - 16.28
- for 10 µM of both sense and antisense and to put it together to equal 5 µM in total...
- signal - sense µl - antisense µl - water µl
- 1a - 32.32 - 27.79 - 38.89
- 2b - 32.40 - 31.85 - 35.75
- 1c - 29.50 - 40.62 - 29.88
- r2 - 23.51 - 30.71 - 45.78
- 2 µl signal, 5 µM, double stranded!
- 4.17 µl switch, 2.49 µM
- 2.5 µl RPO
- 50 µl Paperbuffer
- 10 µl DTT 100 µM
- 5 µl rNTPs
- 26.3 µl H2O
- 3.1 x
- 12.93 µl switch, 176 µM
- 7.75 µl RPO
- 155 µl Paperbuffer
- 31 µl DTT 100 µM
- 15.5 µl rNTPs
- 81.53 µl H2O
- signals: r2, 1a, 1c
- 1x positive control
- 2 µl signal, 5 µM, double stranded!
- 2.94 µl switch, 2.94 µM
- 2.5 µl RPO
- 50 µl Paperbuffer
- 10 µl DTT 100 µM
- 5 µl rNTPs
- 27.57 µl H2O
- signal: r2
- Cloning Malachitegreen-binding aptamer into pB1C3
- E/P digestion
- 2 µl 10x NEB 4
- 2 µl 10x BSA
- 0.5 µl EcoRI
- 0.5 µl PstI
- 15 µl linearized pB1C3 (50 ng/µl)/malachitegreen binding aptamer (203 ng/µl)
- 37°C, 1 h
- purification afterwards using DNA clean and concentrated (or something)
- Concentrations
- backbone: 11 ng/µl
- insert:
- Ligation
- 5 µl digested plasmid
- 2 µl malachitegreen binding aptamer, 1:10 diluted
- 2 µl T4 ligase buffer
- 1 µl T4 ligase
- 10 µl water
- no transformation not possible: no chloramphenicol plates in physic's department...
19.10.2010
- Yesterday's malachitegreen assay
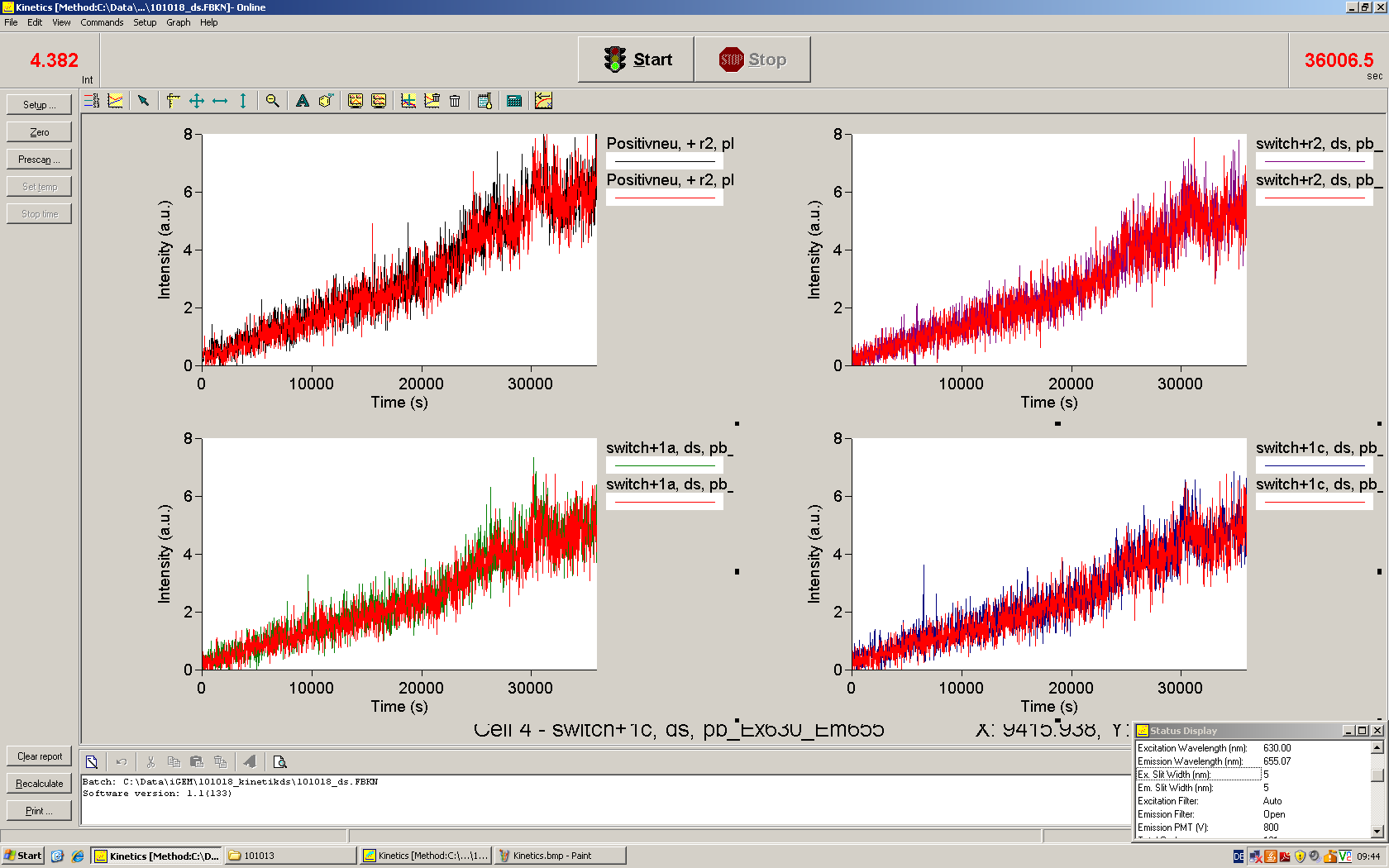
- Emission spectra: Exc=530 nm
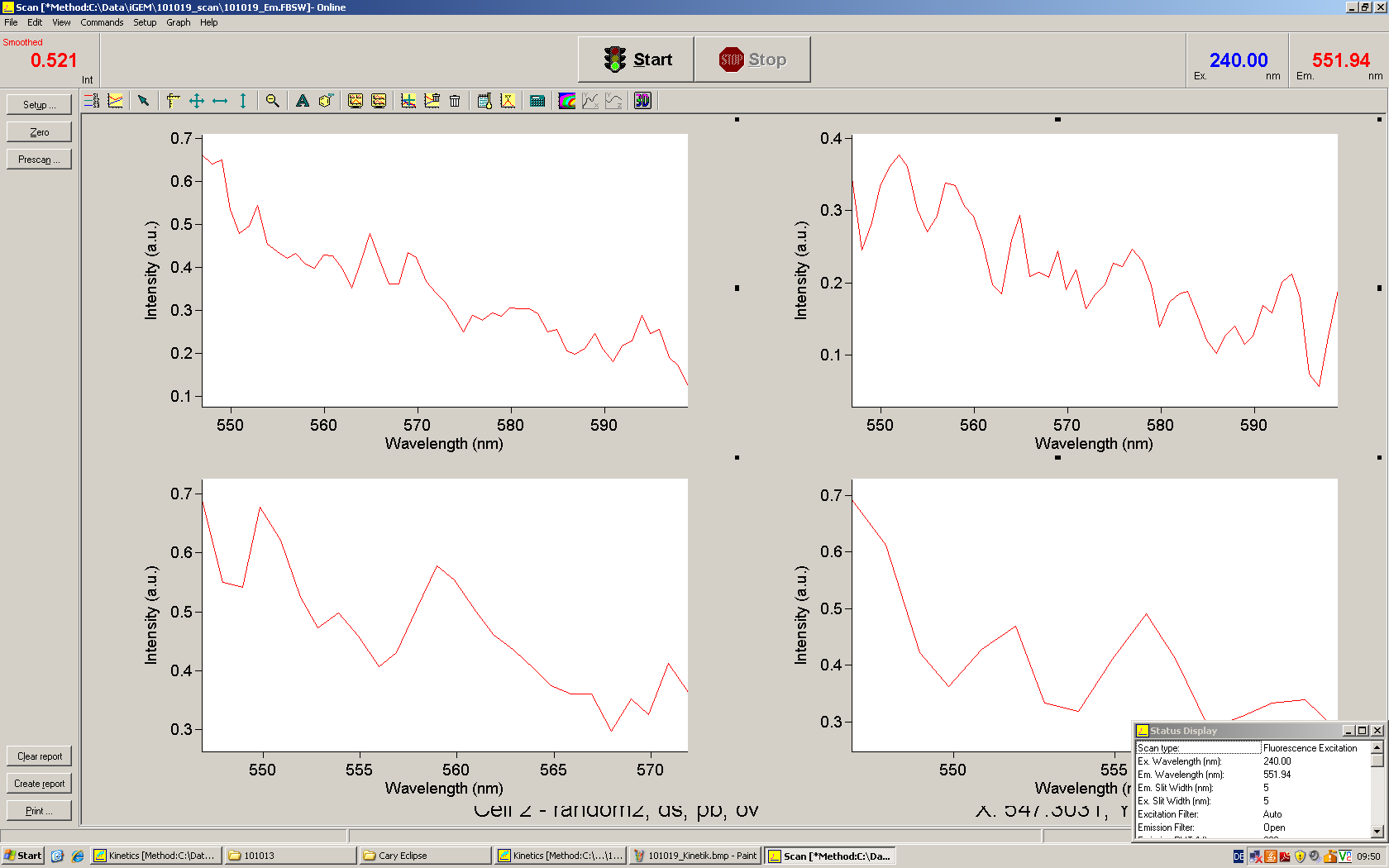
- Excitation spectra: Em=552 nm
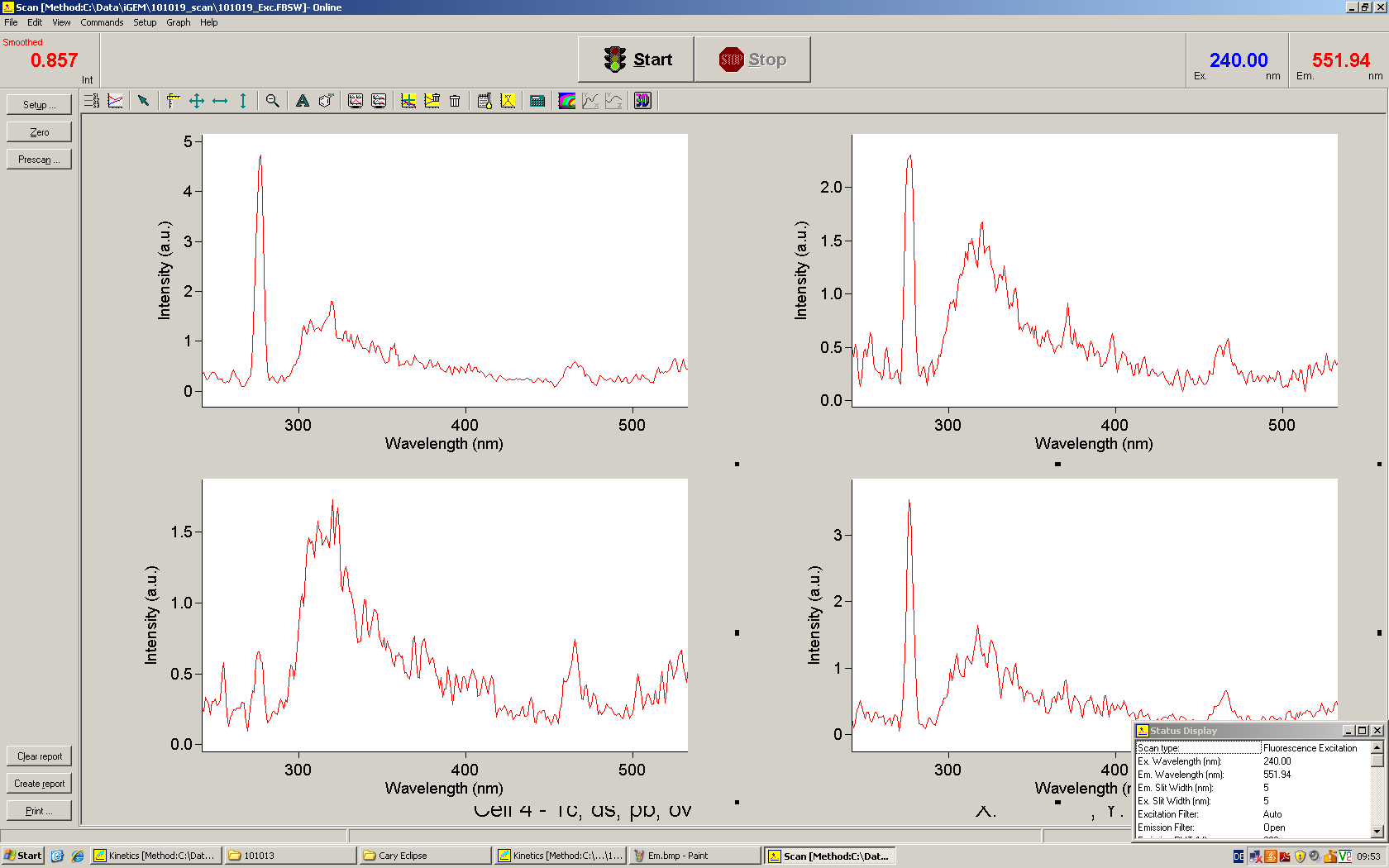
- DnaseI testdigestion
- plasmid digestion to test DnaseI activity:
- 10 µl paper buffer
- 0.2 µl DTT
- 2 µl 10x DnaseI buffer
- 1 µl Dnase
- 1 µl plasmid (some random thing from Wuschel)
- 5.8 µl water
- 37°C, 2h
- samples for DNaseI digestion
- 2 µl 10x DnaseI Buffer
- 1 µl DnaseI
- 17 µl reaction product from malachitegreen assay, 18.10.10
- 37°C, 2 h
- 7M urea, 15 % PAGE
- 30 ml
- 11.25 ml acrylamide
- 12.6 g urea
- 3 ml 10x TBE
- 300 µl 10 % APS
- 30 µl TEMED
- H20 to 30 ml
- heat a bit and sonificate
- gel could not be run: pockets not solid...
- most likely reason: too hot when radical starter were added: instant polymerization were they hit the mixture...
- malachitegreen assay with more malachitegreen this time!
- --> 3x more than usual
- 30 mM 2x malachitegreen paper buffer
- 1 ml 5x paper buffer without malachitegreen
- 0.6 ml 250 mM malachitegreen
- 0.9 ml Water
- 1x switch
- 2 µl signal, 5 µM, double stranded!
- 4.17 µl switch, 2.49 µM
- 2.5 µl RPO
- 50 µl Paperbuffer
- 10 µl DTT 100 µM
- 5 µl rNTPs
- 26.3 µl H2O
- signals: 1a, r2
- 1x positive control
- 2 µl signal, 5 µM, double stranded!
- 2.94 µl switch, 2.94 µM
- 2.5 µl RPO
- 50 µl Paperbuffer
- 10 µl DTT 100 µM
- 5 µl rNTPs
- 27.57 µl H2O
- signal: r2, none
- Cloning Malachitegreen-binding aptamer into pB1C3
- Ligation
- 5 µl digested plasmid
- 2 µl malachitegreen binding aptamer, 1:10 diluted
- 2 µl T4 ligase buffer
- 1 µl T4 ligase
- 10 µl water
- Transformation
- borrowed plates from the Prof. Groll's department
- and from Prof. Becker's
- the one from Prof. Becker's once contained tetracyclin...
- in DH5alpha cells
- 200 µl plated
- overnight, 37°C
20.10.2010
- Yesterday's malachite green binding assay
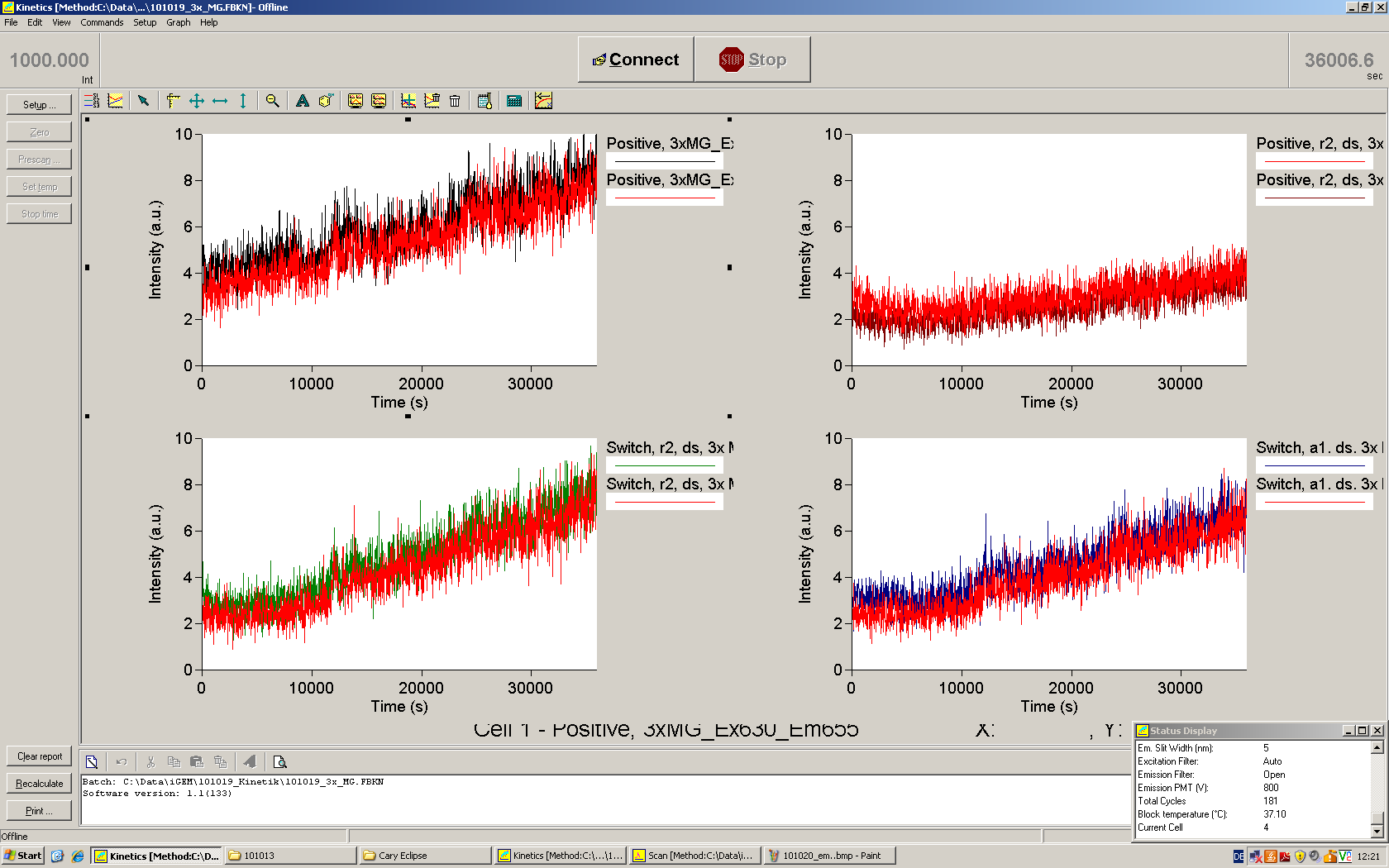
- Emission spectra: Exc=530 nm
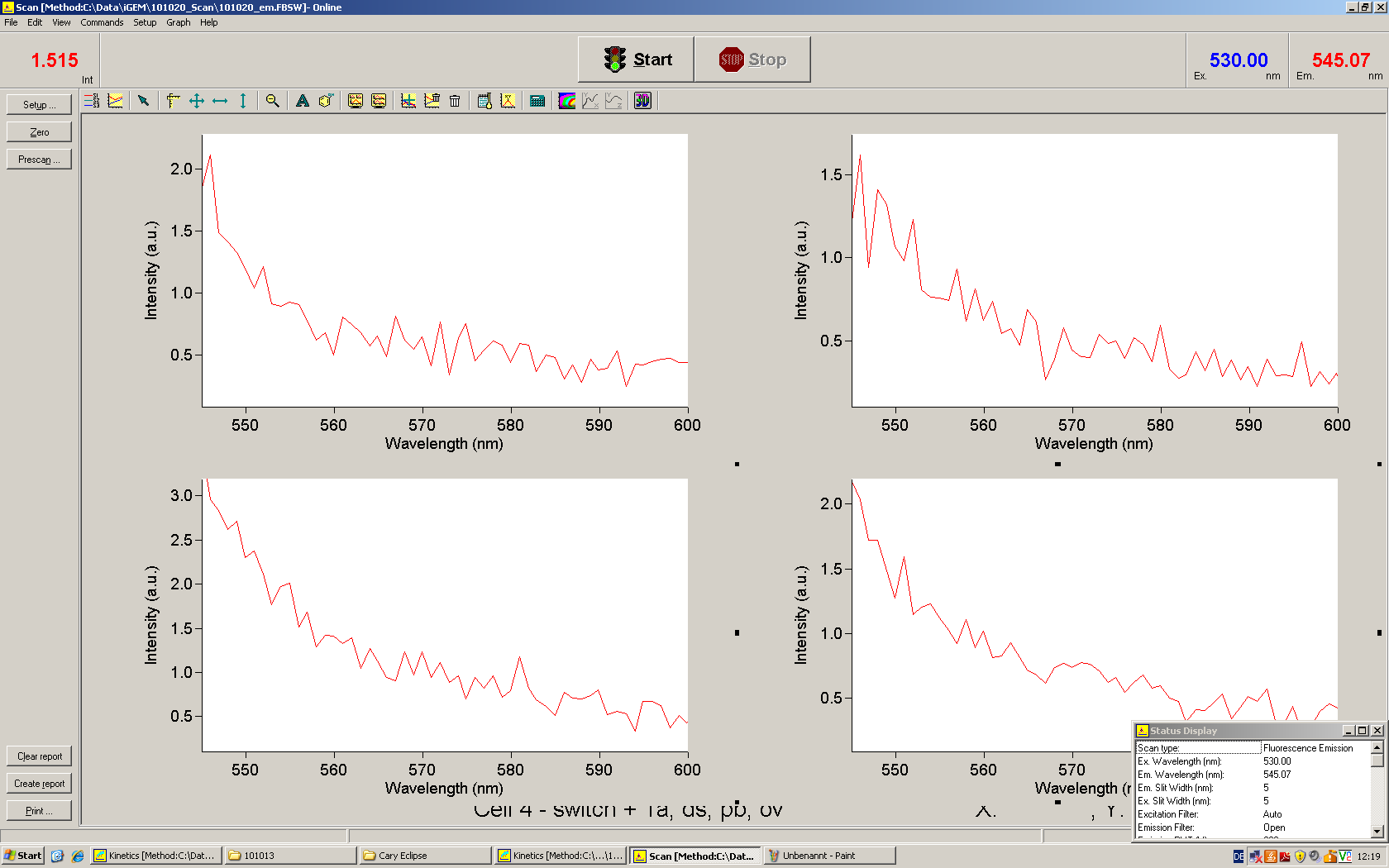
- Excitation spectra: Em=552 nm
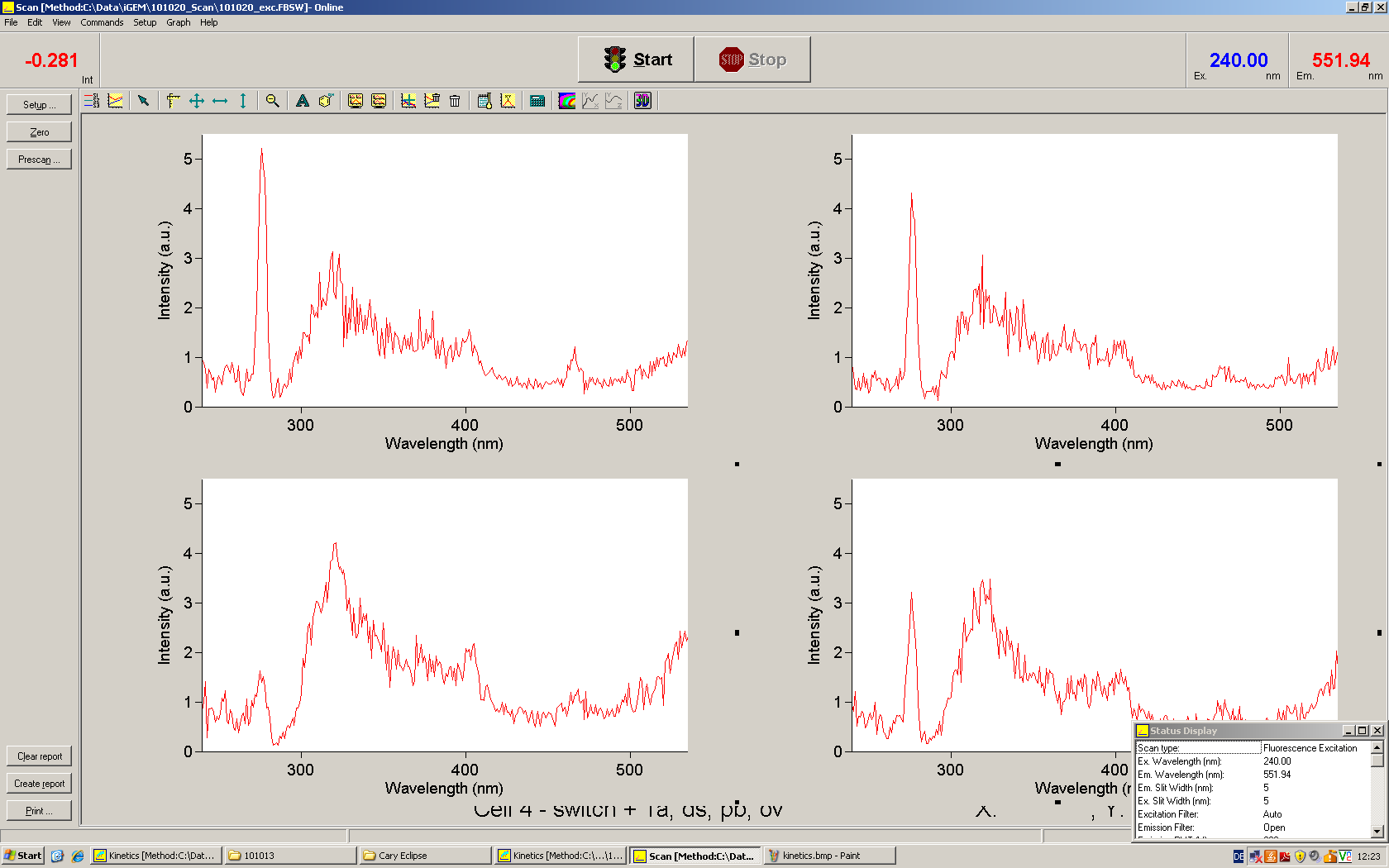
- Cloning Malachitegreen-binding aptamer into pB1C3
- many colonies grown
- Well done, Flo!
- Colony PCRl
- 10 x Mastermix: 500 µl total volume
- 10 µl dNTPs
- 10 µl G1004
- 10 µl G1005
- 50 µl 10x buffer
- 30 µl MgCl2
- 2 µl Taq Polymerase
- 2 µl Template per reaction
- H2O
- PCR program
- iGEM program!
- 58°C annealing
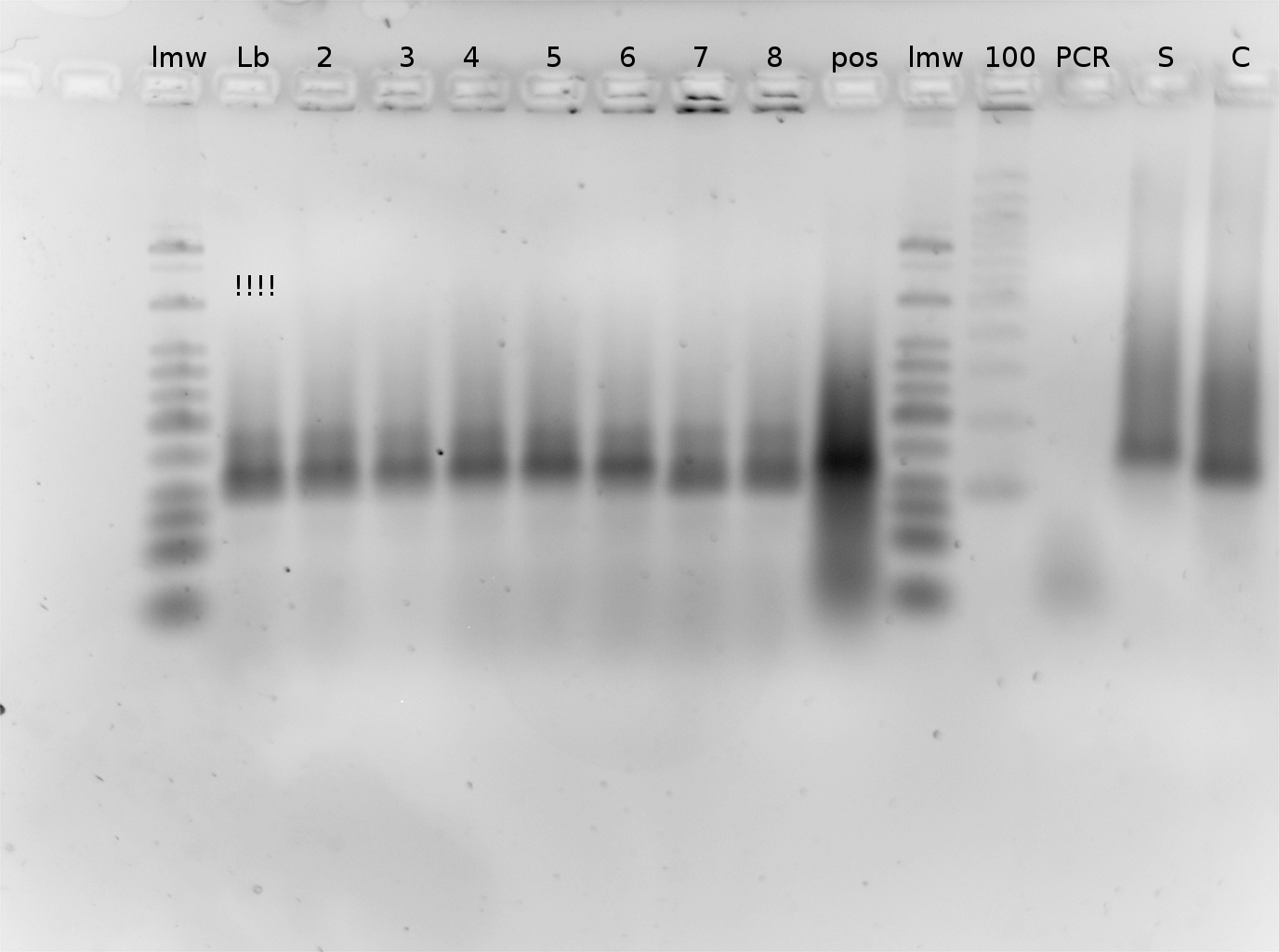
- lmw: low molecular weight (ladder)
- 100: 100 kb (ladder)
- 7 clones chosen to be checked by Colony PCR
- LB=negative control=LB-Chloramphenicol
- positive control: PCR with Biomer product
- also on the gel:
- S=switch=PCR of switch used for measurement
- P=positive control=PCR product used for measurement and cloning
- Interpretation
- stupid negative control looks just the same like everything else
- two bands at approximately the right height, but exactly above and below positive control band
- shit.
- Further proceedings
- 5 ml cultures of clones 3, 4, 5, 7
- 5 ml cultures of 6 new colonies
- minipreps and analytical digestions tomorrow
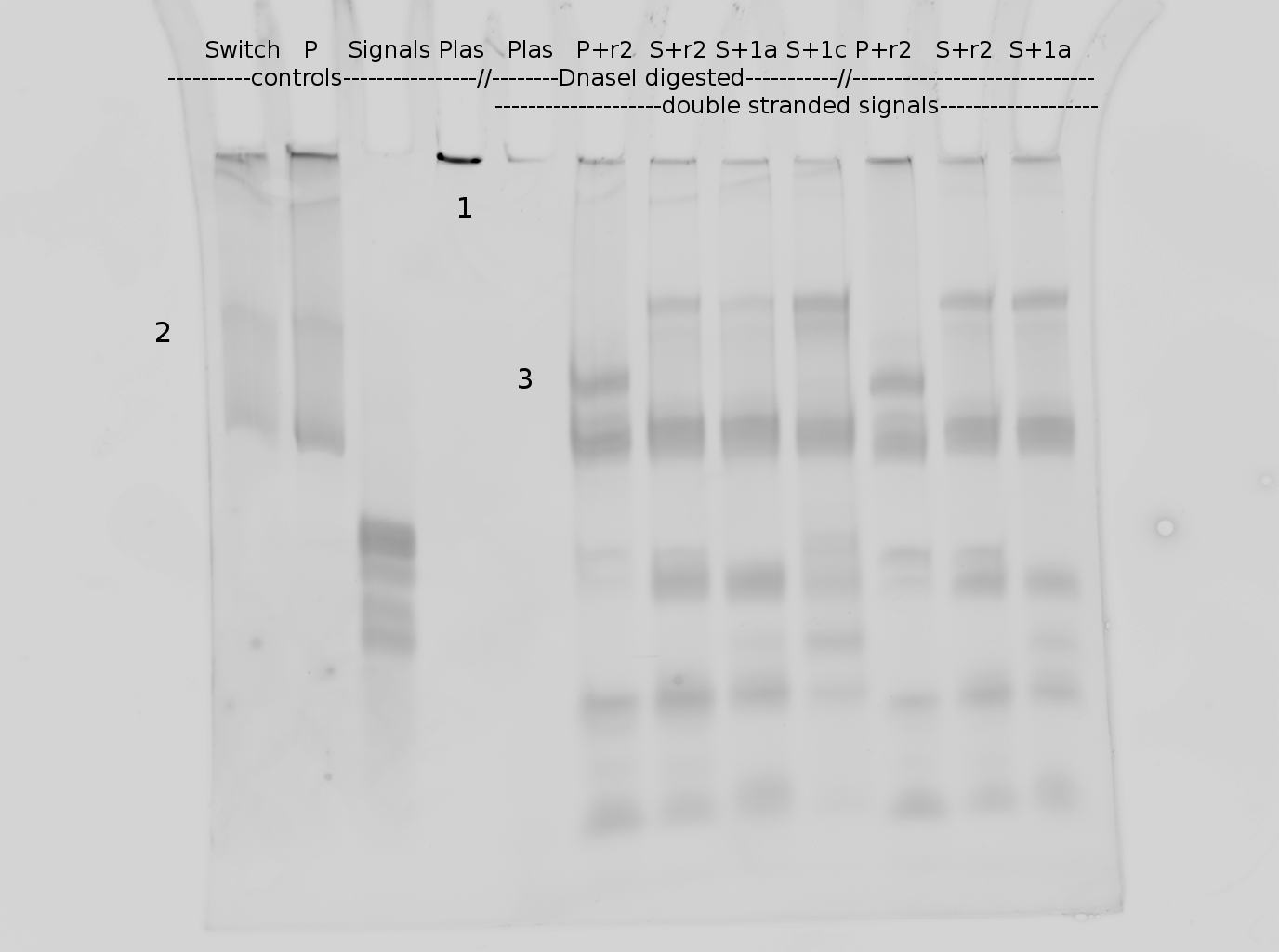
- controls:
- switch: PCR of switch, used in measurements
- P: PCR of positive control, used in measurements
- Plas: random test plasmid used for evaluation of DnaseI activity
- DNase digested samples: 20 µl
- Plas: DnaseI digested random plasmid, for conditions check previous day
- P+r2: Positive control together with double stranded nonsense 2/random 2 signal after overnight transcription in paper buffer (check malachitegreen assay, previous day), DnaseI digested
- S+r2: Switch together with double stranded nonsense 2/random 2 signal after overnight transcription in paper buffer, DnaseI digested
- S+1a: Switch together with double stranded 1a signal after overnight transcription in paper buffer, DnaseI digested
- S+1c: Switch together with double stranded 1c signal after overnight transcription in paper buffer, DnaseI digested
- other: 15 µl
- P+r2: Positive control together with double stranded nonsense 2/random 2 signal after overnight transcription in paper buffer (check malachitegreen assay, previous day)
- S+r2: Switch together with double stranded nonsense 2/random 2 signal after overnight transcription in paper buffer
- S+1a: Switch together with double stranded 1a signal after overnight transcription in paper buffer
- no marker this time! Only 12 lanes :)
- Interpretation:
- no real differences between DnaseI digested and not
- It is rather stupid to use a plasmid to check for DnaseI activity if you want to run the result on a 15 % gel. A plasmid with some kb does not seperate... But: DNA visible, without Dnase I, no DNA visible with DnaseI (stuck in the upper lane, 1)
- Next try with PCR product maybe
- weird lane in all PCR reactions at 2 in both cases and in all other gels and a bit also in agarose gels visible
- positive control runs totally elsewhere than switch even though they do not really vary in length (3)
- Malachitegreen binding assay
- Measurement
- 1x switch
- 2 µl signal, 5 µM, double stranded!
- 4.17 µl switch, 2.49 µM
- 2.5 µl RPO
- 50 µl Paperbuffer
- 10 µl DTT 100 µM
- 5 µl rNTPs
- 26.3 µl H2O
- measured: signals all doublestranded, everything in paper buffer, 10 µM malachitegreen
- positive control
- positive control with nonsense2/random2
- switch with nonsense2/random2
- switch with 1a
21.10.2010
- Fluorescence spetrum, Exc=530
- Fluorescence excitation spectrum, Em= 552
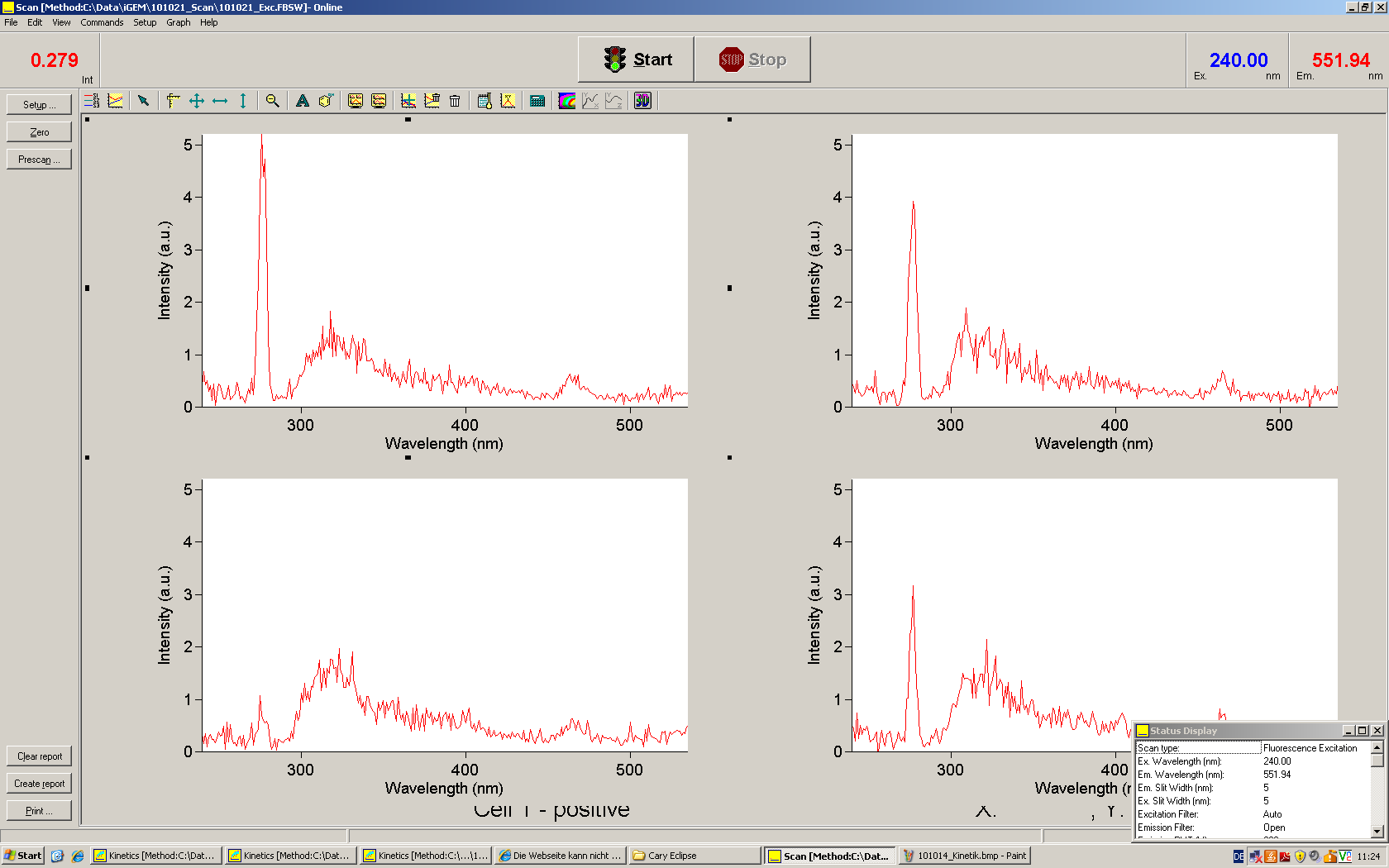
- Amplifying in vivo positive control to send for PartsRegistry
- Miniprep of 5 ml overnight control
- P1 185 ng/µl
- P2 416 ng/µl
- check below for gel picture and further proceeding
- Cloning Malachitegreen-binding aptamer into pSB1C3
- Mini-prep of 5 ml overnight cultures
- # - concentration in ng/µl
- 3 - 272
- 4 - 59
- 5 - 100
- 7 - 95.56
- a - 86
- b - 165
- c - 114
- d - 131
- e - 232
- f - 144
- P1 - 185
- P2 - 416
- Control digestion
- 4 µl plasmid
- 2 µl NEB 3
- 0.5 µl PstI
- 0.5 µl EcoRI-HF
- 13 µl H2O
- 1 h, 37°C
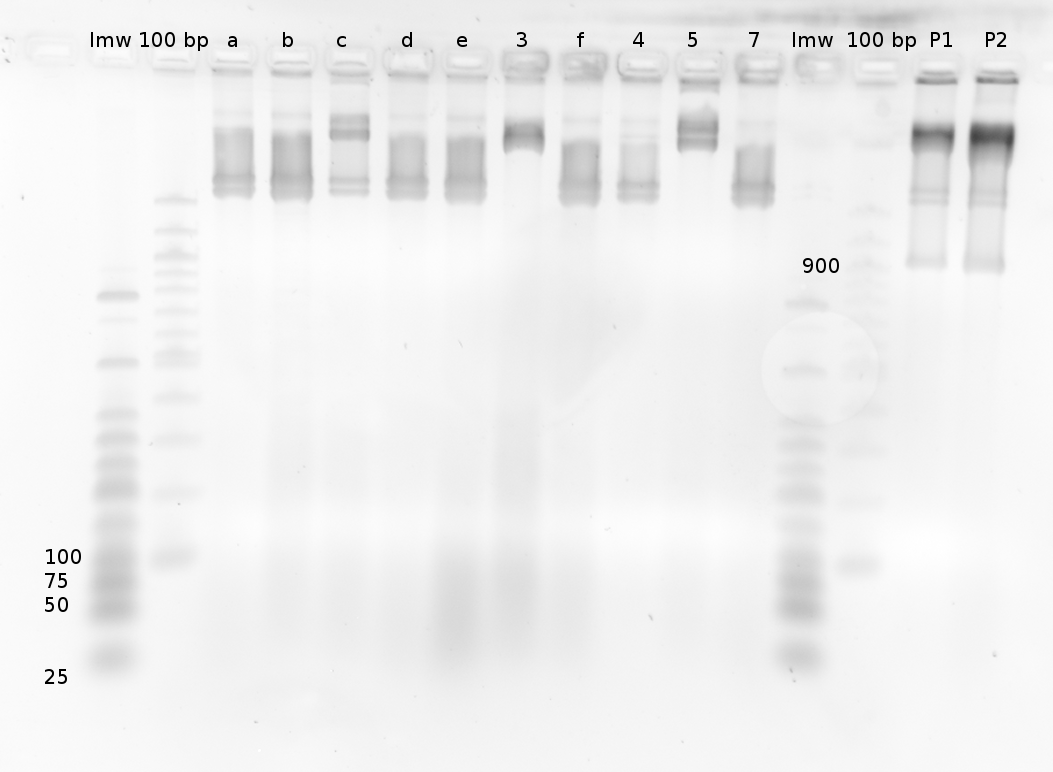
- lmw: low molecular weight ladder (NEB)
- 100 bp: 100 bp ladder (NEB)
- 3,4,5,7: yesterday's clones, checked and falsified by colony PCR
- a-g: yesterday picked clones
- P1, P2: minpreps of in vivo measurement positive control, to be sent to partsregistry
- Interpreation
- well, something went seriously wrong while cloning
- asked at physic's department: DH5alpha cells in use were pretty old: cell from old -80°C are baaaad!
- showed us new stock
- further possibilities: EcoRI nearly empty --> not cutting properly anymore
- T4 ligation buffer: went through 4 tubes until I found one which was not totally full of precipitates
- Chloramphenicol-plates: got plates from Groll group, plates from 2008
- Further cloning procedure
- digestion of linearized pSB1C3 (from parts registry) and positive control with EcoRI-HF (full enzyme)
- digestion of T7 positive control
- 15 µl linearized pSB1C3 (25 ng/µl, damn it, I thought there are 50 ng/µl while preparing the ligation...)/5 µl of positive control, PCR product (c=173 ng/µl) + 10 µl H2O
- 2 µl 10x NEB 3
- 2 µl 10x BSA
- 0.5 µl EcoRI
- 0.5 µl PstI
- 37°C, 1 h
- ligation
- 1 µl T4 ligase
- 2 µl T4 ligase buffer: tested three buffers, one was okay, made aliquots: what happened to all the buffers???
- 1.33 µl backbone (I thought 50 ng, actually 25 ng)
- 0.23 µl insert, 1:10 diluted
- 17.37 µl H2O
- incubated till transformation
- Extracting pSB1C3 from iGEM 2010 distribution
- 10 µl H20 in A3
- 3 µl used for transformation
- transformation
- in DH5alpha cells from new -80°C fridge...
- also in XL-1 blue: Tetracycline resistance
- 15 minutes in ice with ligation product
- 1 minute heat shock, 42°C
- 2 minutes on ice
- 500 µl LB0 added
- 1.5 hours at 37°C, shaking
- control: digested, linearized pSB1C3 into XL-1 blue
- reason: DpnI digestion recommended, we don't have any DpnI
- on chloramphenicol agar plates from Prof. Buchner's lab --> new and shiny
- XL-1 blue plated on Tetracycline/Chloramphenicol plates from Becker's lab: not used in Becker's lab because Tetracycline broken, does not matter for us...
- E. coli RPO malachitegreen assay
- E. coli RPO finally arrived
- called Biozym: RPO was supposed to arrive yesterday, delivery cimpany signed for yesterday
- called Materialausgabe two times everyday for the last week --> In the end they already knew, when they heard E14
- nevertheless when the enzyme arrived, somebody forgot to put it into the large book of incoming stuff
- only one person knew, that the enzyme already arrived
- stored at -20 °C meantime
- dry ice already vaporated...
- people were first mad at me, because I insisted that the package arrived although it was not written in the main book
- then mad at each other because somebody did not put it into the main book
- then I was slightly mad because we called two times a day for seven days, argued about -80°C storage and still...
- bought dry ice, transport into ZNN, stored at -20°C covered in dry ice (-80°C fridge is already standing in the lab but not installed yet) until measurements
- now stored at old -80°C in physic's department in iGEM box
- preperation of measuring constructs - without terminator
- PCR of 12z (His-Terminator in plasmid)
- 5z (TrpTerm in plasmid)
- 28z (pSB1A2-R0011-TrpSig)
- pSB1K3-R0011-HisSig-B0014
- 4x 8x PCR mix
- 8 µl dNTPs
- 8 µl Apt forward
- 8 µl Apt reverse - without terminator!
- 40 µl 10x buffer
- 24 µl MgCl2
- 1.6 µl Taq Polymerase
- 302.4 µl Template per reaction
- H2O
- recognized that wrong primers were used for the signals, thrown away after PCR
- protocol iGEM PCR, 52°C annealing temp.
- purified using Clean and Concentrator
- yields: 220 ng/µl TrpTerm, 178 ng/µl (or something like that) HisTerm
- Buffer preparation for E. coli RPO
- Instructions from Epicentre Biotechnologies Datasheet (included in package)
- 5x transcription buffer
- 2 ml Tris, pH=7.49
- 1 ml 500 mM MgCl2
- 5 µl Triton X-100
- 3.75 ml 2 M KCl
- water to 10 ml
- stored in fridge
- 2x transcription buffer with malachitegreen
- 4 ml 5x buffer
- 800 µl 250 mM malachitegreen stock
- water to 10 ml
- wrapped in aluminium foil, stored in fridge
- measurement with E. coli RNA Polymerase
- measured: 12z (TrpTerm - negative control), 5z (HisTerm, negative control), (positive control)
- 1x measurement
- 2.5 µl RPO (2.5 U)
- 10 µl DTT (100 mM stock, 10 mM end)
- 50 µl 2x buffer
- 2.5 µl rNTPs (80 mM stock, 2 mM end)
- 1 µg DNA template
- water to 100 µl
- 3.1 x
- 7.75 µl RPO
- 31 µl DTT
- 7.75 µl rNTPs
- 155 µl 2x buffer
- 77.5µl H20
- 4.5 µl 5z/5.5 µl H2O
- 5.35 µl 12z/4.65 µl H2O
- 5.88 µl ??? /4.12 µl H20
- 38°C, Exc=630 nm, Em=650/655 nm
- Cloning of R0011 and B1006 in pSB1K3
- digestion of R0011 with EcoRi and SpeI (check above, same procedure)
- ligation
- 0.3 µl R0011
- 0.2 µl B1006 (signal)
- 5 µl pSB1K3
- 2 µl T4 ligation buffer
- 1 µl T4 ligase
- 11.5 µl H2O
- Transformation into DH5alpha cells
- put on Kana-Plates
- Cloning of signal B1006 in pSB1A2_R0011
- 1.82 µl pSB1A2_R0011
- 0.2 µl Signal B1006
- 2 µl T4 DNA ligation buffer
- 1 µl T4 DNA ligase
- 15 µl H2O
- Transformation into DH5alpha cells
- put on Amp-plates
22.10.2010
- Yesterday's malachitegreen assay using E. coli RPO
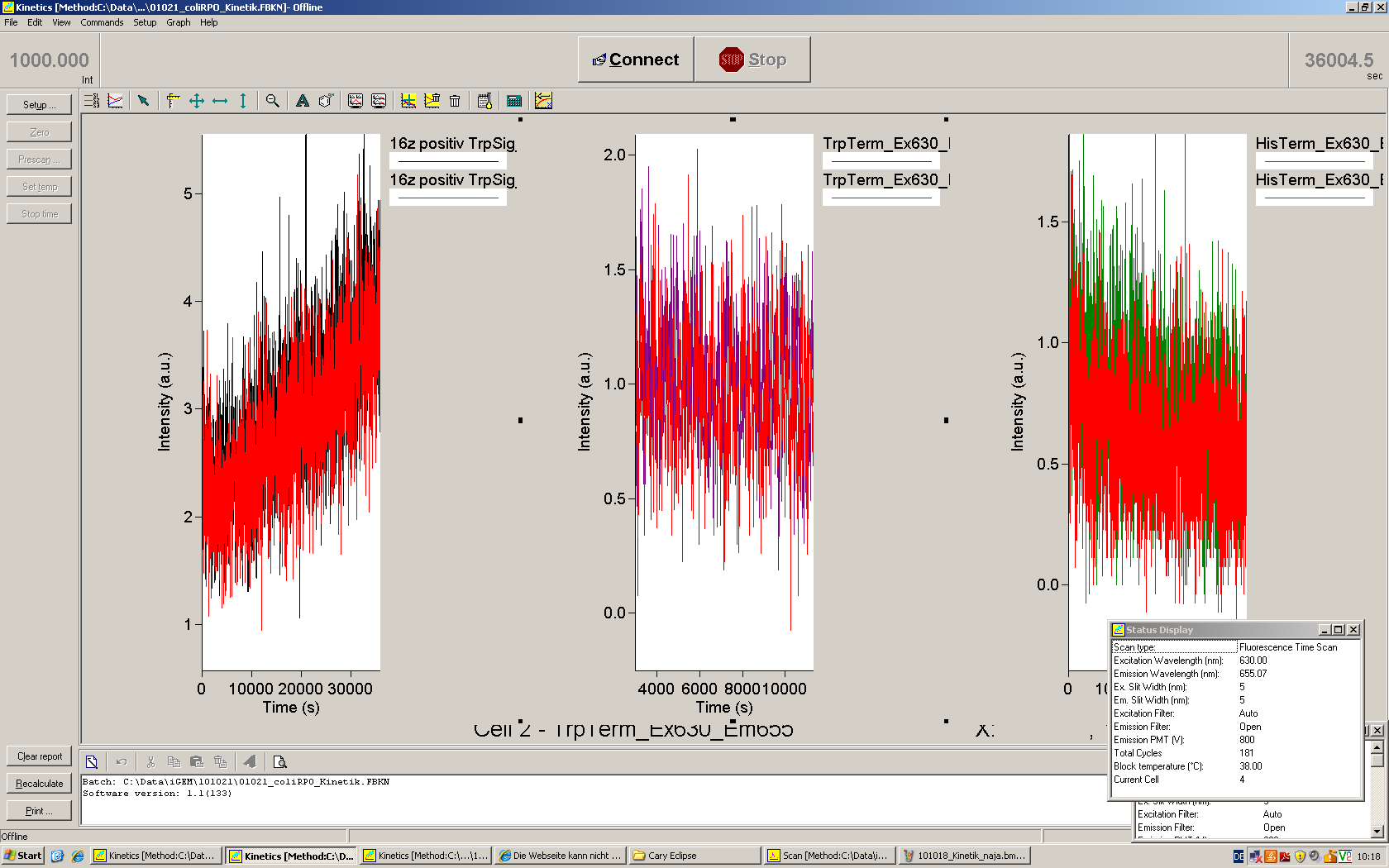
- Fluorescence spetrum, Exc=630
- Fluorescence excitation spectrum, Em= 652
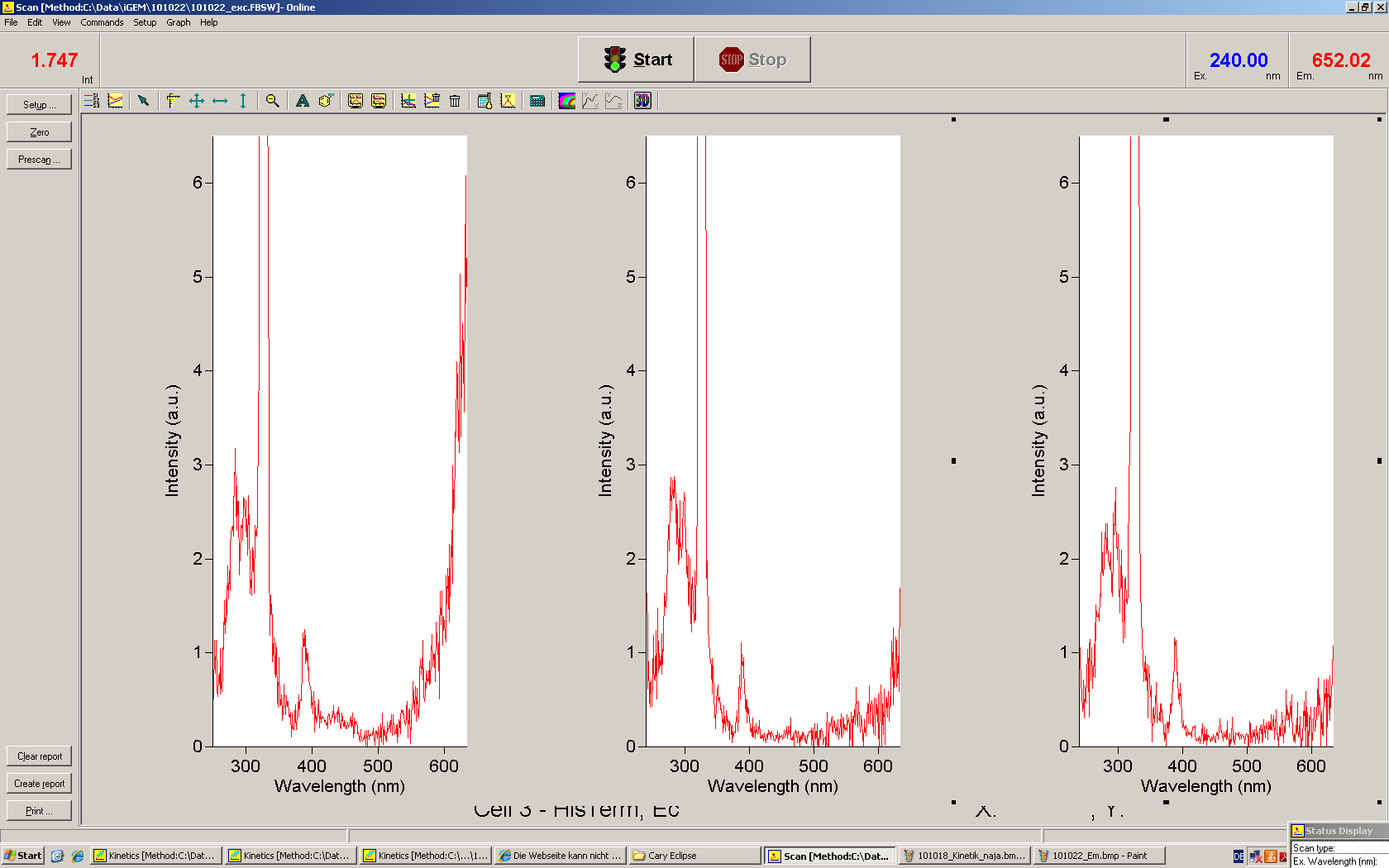
- 7 M 15 % PAGE
- DnaseI digestion of E. coli RPO transcription
- control: T7 positive control
- 17 µl Transcription product
- 2 µl 10 x DnaseI buffer
- 1 µl Dnase
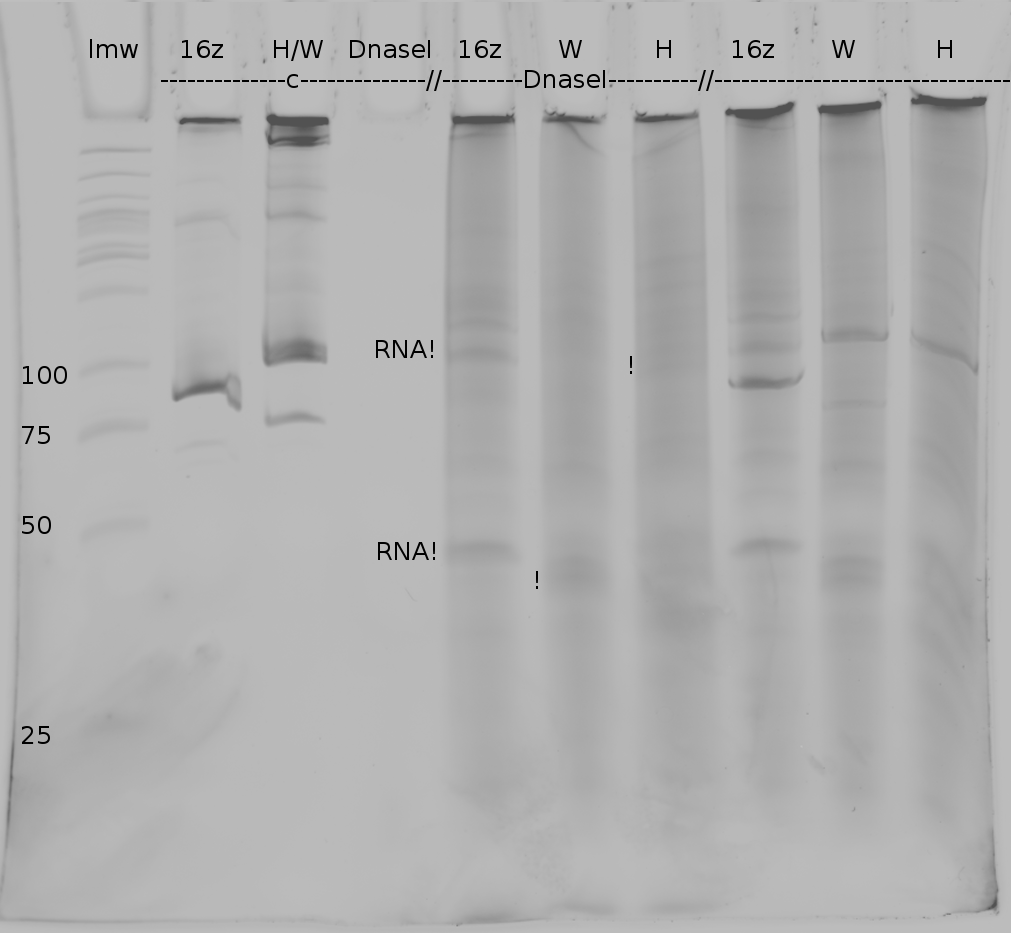
- lmw: low molecular weight ladder, NEB
- H: His-switch
- W: Trp-Switch
- 16z: Positive control (with TrpSignal); using Primer Apt_For+AptPart_woT_Rev
- Ladies and Gentleman, we definetly see RNA this time
- positive control completely transcriped, terminators terminate (finally some terminator terminate...)
- DnaseI digestion works. DnaseI control completely clean.
- I think termination products are visible
- I also think, that RPO/DnaseI/protein in general bound RNA is stuck in pockets
- 1h. 37°C, Dnase digestion conditions: RNA suffers a bit, paradise for RNases?
- Cloning Malachitegreen-binding aptamer into pSB1C3
- no clones on plates
- clones expected to show up on A3 transformed cells --> part from partsregistry!
- weird clones on Tetracycline/Chloramphenicol plates from Becker's department --> maybe both antibiotics not working anymore
- talked to Moni from E 22
- still old cells in new -80°C --> cells from 2008, maybe dead, certainly not competent...
- repeat transformation (see yesterday's labbook)
23.10.2010
- In vitro transcription using E. coli RPO
- PCR of HisSig, TrpSig, HisTerm, TrpTerm, 16z (positive control)
- 100 µl per template, 800 µl total
- 16 µl dNTPs
- 16 µl forward primer
- 16 µl reverse primer
- 80 µl Taq-buffer Mg-free
- 3.2 µl Taq Polymerase
- 48 µl MgCl2
- 5 µl template per 100 µl
- 615.8 µl H2O
- PCR Purification using DNA Quick and Clean
- yield: Template - # - concentration [ng/µl]
- 16z - 1 - 146
- 16z - 2 - 138
- 16z - 3 - 138
- 16z - 4 - 128
- HisTerm - 1 - 96
- HisTerm - 2 - 160
- TrpTerm - 1 - 204
- TrpTerm - 2 - 190
- HisSig - 1 - 160
- HisSig - 2 - 165
- TrpSig - 1 - 83.5
- TrpSig - 2 - 166
- Measurement
- 16z positive control, TrpTerm, both with and without signal
- 4.1 x Master Mix
- 105 µl Buffer
- 41 µl DTT
- 10.25 µl RPO
- 10.25 µl rNTPs
- 77.9 µl H2O
- take 84 µl Master Mix
- add to:
- 10.85 µl 16 z + 6.1 µl H20
- 10.85 µl 16 z + 6.1 µl TrpSig
- 10.92 µl TrpTerm + 6.1 µl H20
- 10.92 µl TrpTerm + 6.1 µl TrpSig
600px
Only the positive control without Signal increases!
- Cloning of MPA into pSB1C3
- Checked plates, no clones
- being sad
- said stupid plates
- Repetition of ligation
- for 50 ng plasmid backbone
- 2.6 µl pBS1C3 E/P digested
- 0.23 µl MPA, E/P digested
- 2 µl T4 ligase buffer - I checked four buffers, all were precipitated, took a completely new one and aliquoted it!
- 1 µl T4 Ligase
- for 100 ng plasmid backbone
- 5.2 µl pBS1C3 E/P digested
- 0.46 µl MPA, E/P digested
- 2 µl T4 ligase buffer - I checked four buffers, all were precipitated, took a completely new one and aliquoted it!
- 1 µl T4 Ligase
- transformation into pSB1K3 (just in case...)
- 4.5 µl pSB1K3 E/P digested
- 0.23 µl MPA, E/P digested
- 2 µl T4 ligase buffer - I checked four buffers, all were precipitated, took a completely new one and aliquoted it!
- 1 µl T4 Ligase
- found some pSB1C3_RFP clones in the evening, 5 ml cultures
- stroke over the plate with a pipette tip, 5 ml culture
24.10.2010
- Cloning of MPA into pSB1C3
- Miniprep of pSB1C3_RFP and something random from the plate of no clones
- mixed up samples: yields were good, but I guess they don't matter
- very tiny clones found in the morning
- slightly bigger ones by noon
- pickable clones by evening
- picked 10 clones, 5 ml cultures
- BBa_K494001-BBa_K494006
- 2x 5 ml LB_Amp from glycerin stocks
- Measurements with T7 RPO and E. coli constructs
- 16z positive control, TrpTerm, both with and without signal
- 4.1 x Master Mix
- 205 µl Buffer
- 41 µl DTT
- 10.25 µl RPO
- 10.25 µl rNTPs
- 77.9 µl H2O
- take 76.2 µl Master Mix
- add to:
- 10.07 µl 16 z + 13.7 µl H20
- 10.07 µl 16 z + 9.61 µl TrpSig + 4 µl H2O
- 23.67 µl HisTerm (1)
- 14.2 µl HisTerm (2) + 9.61 µl TrpSig
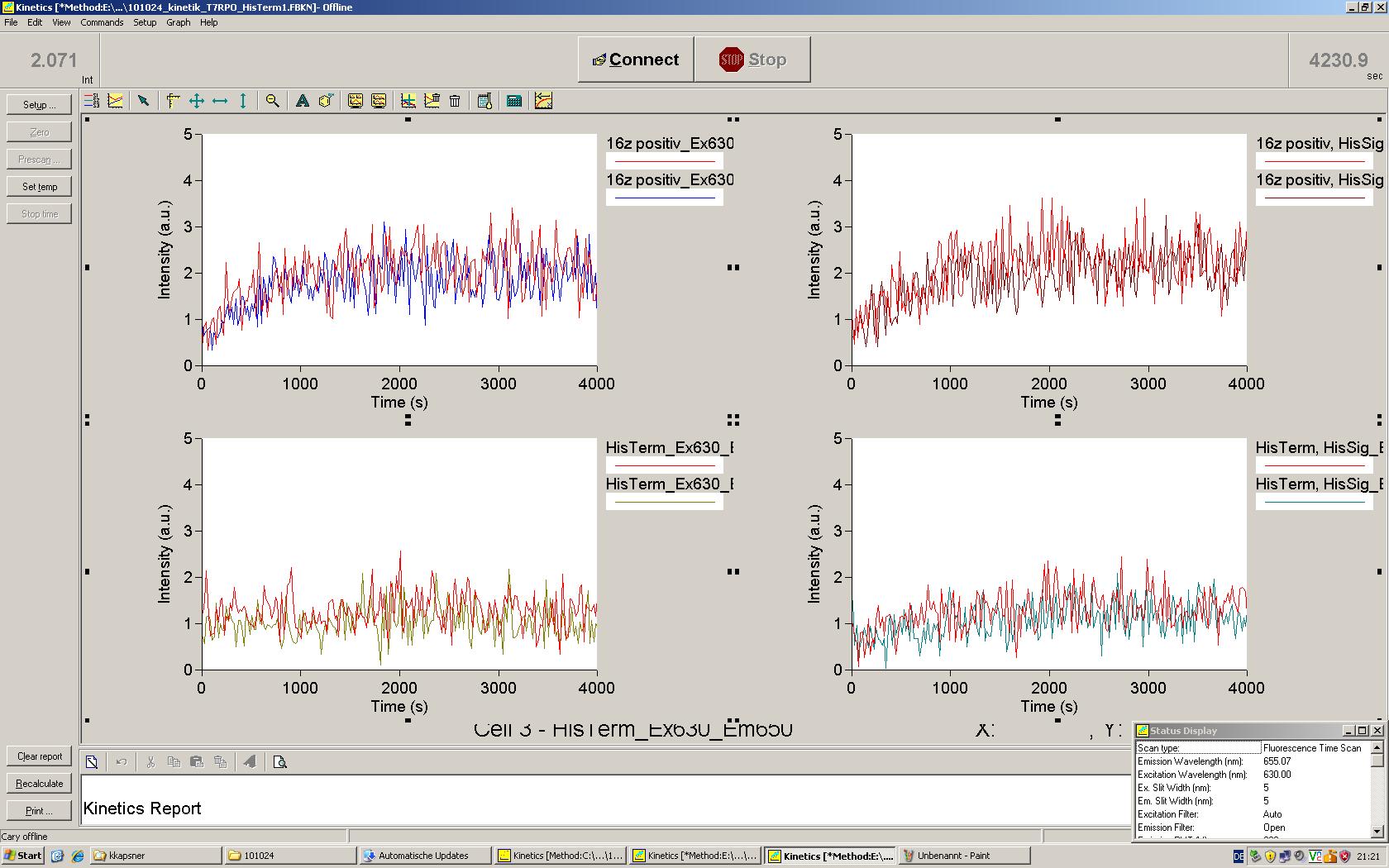 It looks like an increase in 3 of 4 traces, but spectr show no sign of malachite green binding:
It looks like an increase in 3 of 4 traces, but spectr show no sign of malachite green binding:
Close
Week31
Cloning of Parts into pSB1C3 T7 & E. coli
The Week of Wiki freeze!
25.10.2010
- Submitting Parts
- Miniprep of yesterday's 5 ml cultures using Zymo Classic
- MPA into pSB1C3
- # - concentration (ng/µl)
- 1 - 45.5
- 2 - 96.5
- 3 - 19.5
- 4 - 31
- 5 - 122
- 6 - 164
- 7 - 30
- 8 - 39.5
- 9 - 20.5
- 10 - 113
- BBa_K494001-BBa_K494006
- BioBrick - # - concentration in ng/µl
- BBa_K494001 - 1 - 179
- BBa_K494001 - 2 - 182
- BBa_K494002 - 1 - 214
- BBa_K494002 - 2 - 288
- BBa_K494003 - 1 - 160
- BBa_K494003 - 2 - 202
- BBa_K494004 - 1 - 308
- BBa_K494004 - 2 - 322
- BBa_K494005 - 1 - 290
- BBa_K494005 - 2 - 126
- BBa_K494006 - 1 - 290
- BBa_K494006 - 2 - 232
- control digestion of everything
- EcoRI/PstI
- MPA clones 1, 3, 4, 7, 8, 9: 8 µl
- all other: 4 µl
- 7 x for 4 µl
- 14 µl 10 x BSA
- 14 µl 10 NEB3
- 2.1 µl EcoRI HF
- 2.1 µl PstI
- water as needed
- 19 x for 4 µl
- 38 µl 10x BSA
- 38 µl 10x NEB3
- 5.7 µl EcoRI HF
- 5.7 µl PstI
- water as needed
- 37°C, 1 hour, fitted just perfectly into the heat block
- recognized later that MPA 2, 5, 6, 10 were digested without DNA
- NotI Digestion with --> PstI empty... Time to finish :)
- 10 µl NEB
- 10 µl 10x BSA
- NotI
- pSB1C3_MPA (BBa_K494000) run on 2 % agarose gel
- --> clone 1 picked for submission: Looked better in reality!
- BBa_K494001-BBa_K494006 run on 1.5 % agarose gel
- pSB1C3_MPA (BBa_K494000) missed preps run on 2 % agarose gel
26.10.2010
- Waited for the FedEx man from 8:00-15:00. Called at 15:15. Said, oh, they forgot to pick it up! Sent somebody immediately who arrived at 15:45. Happyness --> Parts submitted, not our fault anymore :) Track: 871353522440
27.10.2010
- Ran around to shoot weird E. coli pics
- Not in the lab anymore... FedEx delivered our parts! 9.14 am EDT, we recognized it at 16:22 MEST! More Happyness, now back to serious working maybe...
Close
Protocols
Molecular Biology
Read more
Read more
Taq Polymerase Hot Start
PCR Pippeting plan:
1 µl template
1 µl dNTP 10 µM
1 µl G1004 (Primer) 10 µM
1 µl G1005 (Primer) 10 µM
5 µl 10x Taq-buffer (500 mM KCl, 100 mM Tris-HCl (pH 8.3), 15 mM MgCl2)
0,2 µl Taq-Polymerase (add last) 5,000 U/ml
40.8 µl Water
Final volume 50µl
Processing: (program saved as IGEMPCR )
- preheating of PCR chamber to 94 °C
--> insert sample
- 30 sat 94°C (according to IGEM protocols)
- 30 s at 56 °C
- 45s at 72°C
- 7 min at 72°C
- stay at 4°C
colony PCR
- Colony PCR
- pick colonies and resuspend them in 20 µl LB+Antibiotic (each)
- PCR of 2 µl of each sample, 2 µl as negative control (Program: ColonyPCR, modified), store remaining 18 µl for overnight cultures
- afterwards, mix 15 µl of each PCR product with 3 µl GLPn and load to Gel
- make overnight cultures of positive clones by adding the remaining 18 µl to 5 ml LB+AB
program:colonypcr
- preheating of PCR chamber to 94 °C
--> insert sample
- 5 min 30 sec at 94 °C
- loop 35x:
- 30 sat 94°C (according to IGEM protocols)
- 30 s at 58 °C
- 60s at 72°C
- 7 min at 72°C
- stay at 4°C
Close
Read more
PCR samples
ZYMO RESEARCH DNA Clean&Concentration Kit
[http://www.zymoresearch.com/zrc/pdf/D4003i.pdf Protocol and Information]
- In a 1.5 ml microcentrifuge tube, add 2-7 volumes of DNA Binding Buffer to each volume of DNA sample (see table below). Mix briefly by vortexing.
| Application
| DNA Binding Buffer : Sample
| Example
|
| Plasmid, genomic DNA (>2 kb)
| 2 : 1
| 200 μl : 100 μl
|
| PCR, cDNA, DNA fragment
| 5 : 1
| 500 μl : 100 μl
|
| ssDNA (e.g., M13 phage)
| 7 : 1
| 700 μl : 100 μl
|
- Transfer mixture to a provided Zymo-Spin™ Column1 in a Collection Tube.
- Centrifuge at ≥10,000 x g for 30 seconds. Discard the flow-through.
- Add 200 μl Wash Buffer to the column. Centrifuge at ≥10,000 x g for 30 seconds. Repeat wash step.
- Add ≥6 μl water2,3 directly to the column matrix. Transfer the column to a 1.5 ml microcentrifuge tube and centrifuge at ≥10,000 x g for 30 seconds to elute the DNA.
Ultra-pure DNA in water is now ready for use.
QIAquick purification Kit
[http://www1.qiagen.com/literature/render.aspx?id=103715 Handbook]
Procedure
1. Add 5 volumes of Buffer PB to 1 volume of the PCR sample and mix. It is not necessary to remove mineral oil or kerosene. For example, add 500 μl of Buffer PB to 100 μl PCR sample (not including oil).
2. If pH indicator I has beein added to Buffer PB, check that the color of the mixture is yellow. If the color of the mixture is orange or violet, add 10 μl of 3 M sodium acetate, pH 5.0, and mix. The color of the mixture will turn to yellow.
3. Place a QIAquick spin column in a provided 2 ml collection tube.
4. To bind DNA, apply the sample to the QIAquick column and centrifuge for 30–60 s. We changed it to 3 min @ 6000rpm !
5. Discard flow-through. Place the QIAquick column back into the same tube. Collection tubes are re-used to reduce plastic waste.
6. To wash, add 0.75 ml Buffer PE to the QIAquick column and centrifuge for 30–60 s.
7. Discard flow-through and place the QIAquick column back in the same tube. Centrifuge the column for an additional 1 min.repeat!
IMPORTANT: Residual ethanol from Buffer PE will not be completely removed unless the flow-through is discarded before this additional centrifugation.
8. Place QIAquick column in a clean 1.5 ml microcentrifuge tube.
9. To elute DNA, add 50 μl Buffer EB (10 mM Tris·Cl, pH 8.5) or water (pH 7.0–8.5) to the center of the QIAquick membrane and centrifuge the column for 1 min. Alternatively, for increased DNA concentration, add 30 μl elution buffer to the center of the QIAquick membrane, let the column stand for 1 min, and then centrifuge.
IMPORTANT: Ensure that the elution buffer is dispensed directly onto the QIAquick membrane for complete elution of bound DNA. The average eluate volume is 48 μl from 50 μl elution buffer volume, and 28 μl from 30 μl elution buffer. Elution efficiency is dependent on pH. The maximum elution efficiency is achieved between pH 7.0 and 8.5. When using water, make sure that the pH value is within this range, and store DNA at –20°C as DNA may degrade in the absence of a buffering agent. The purified DNA can also be eluted in TE buffer (10 mM Tris·Cl, 1 mM EDTA, pH 8.0), but the EDTA may inhibit subsequent enzymatic reactions.
10. If the purified DNA is to be analyzed on a gel, add 1 volume of Loading Dye to 5 volumes of purified DNA. Mix the solution by pipetting up and down before loading the gel.
Gel samples
ZYMO RESEARCH Gel DNA Recovery Kit
[http://www.acgtinc.com/PDF_files/Sample%20Prepation_ACGT/Zymoclean%20Gel%20DNA%20Recovery%20Kit_Zymo%20Research.pdf Product informartion]
Protocol
- Excise the DNA fragment1 from the agarose gel using a razor blade or scalpel and transfer it to a 1.5 ml microcentrifuge tube.
- Add 3 volumes of ADB to each volume of agarose excised from the gel (e.g. for 100 μl (mg) of agarose gel slice add 300 μl of ADB).
- Incubate at 37-55 °C for 5-10 minutes until the gel slice is completely dissolved2. For DNA fragments >8 kb, following the incubation step, add one additional volume (equal to that of the gel slice) of water to the mixture for better DNA recovery (e.g. 100 μl agarose, 300 μl ADB and 100 μl water).
- Transfer the melted agarose solution to a Zymo-SpinTM I Column in a Collection Tube.
- Centrifuge at ≥10,000 x g for 30-60 seconds. Discard the flow-through.
- Add 200 μl of Wash Buffer to the column and centrifuge at ≥10,000 x g for 30 seconds. Discard the flow-through. Repeat the wash step.
- Add ≥6 μl of water3,4 directly to the column matrix. Place column into a 1.5 ml tube and centrifuge ≥10,000 x g for 30-60 seconds to elute DNA.
Ultra-pure DNA in water is now ready for use.
Miniprep
Protocol:
- Add 600 μl of bacterial culture grown in LB medium to a 1.5 ml microcentrifuge tube.
- Add 100 μl of 7X Lysis Buffer (Blue)1 and mix by inverting the tube 4-6 times. Proceed to step 3 within 2 minutes. After addition of 7X Lysis Buffer the solution should change from opaque to clear blue, indicating complete lysis.
- Add 350 μl of cold Neutralization Buffer (Yellow)2 and mix thoroughly. The sample will turn yellow when the neutralization is complete and a yellowish precipitate will form. Invert the sample an additional 2-3 times to ensure complete neutralization.
- Centrifuge at 11,000 – 16,000 x g for 2-4 minutes.
- Transfer the supernatant (~900 μl) into the provided Zymo-Spin™ IIN column. Avoid disturbing the cell debris pellet.
- Place the column into a Collection Tube and centrifuge for 15 seconds.
- Discard the flow-through and place the column back into the same Collection Tube.
- Add 200 μl of Endo-Wash Buffer to the column. Centrifuge for 15 seconds. It is not necessary to empty the collection tube.
- Add 400 μl of Zyppy™ Wash Buffer2 to the column. Centrifuge for 30 seconds.
- Transfer the column into a clean 1.5 ml microcentrifuge tube then add 30 μl of Zyppy™ Elution Buffer3 directly to the column matrix and let stand for one minute at room temperature.
- Centrifuge for 15 seconds to elute the plasmid DNA.
Close
Read more
Restriction Digest
Enzyme
| 10 units is sufficient, generally 1µl is used
|
| DNA
| 1 µg
|
10X NEBuffer
| 5 µl (1X)
|
| BSA
| Add to a final concentration of 100 µg/ml (1X) if necessary
|
| Total Reaction Volume
| 50 µl
|
| Incubation Time
| 1 - 1.5 hour
|
| Incubation Temperature Enzyme dependent
|
XbaI, SpeI, PstI, SpeI : 37 °C
|
[http://www.neb.com/nebecomm/tech_reference/restriction_enzymes/buffer_activity_restriction_enzymes.asp activity of restriction enzymes in NEB buffers]
Biobrick standard
[http://openwetware.org/wiki/Restriction_digest Protocols for IGEM standard digestion]
Dephosphorylation
using Antarctic Phosphatase
- Add 1/10 volume of 10X Antarctic Phosphatase Reaction Buffer to 1-5 µg of DNA cut with any restriction endonuclease in any buffer.
- Add 1 µl of Antarctic Phosphatase (5 units) and mix.
- Incubate for 15 minutes at 37°C for 5´ extensions or blunt-ends, 60 minutes for 3´ extensions.
- Heat inactivate (or as required to inactivate the restriction enzyme) for 5 minutes at 65°C.
- Proceed with ligation.
[http://www.neb.com/nebecomm/products/protocol76.asp from NEB]
Close
Read more
Using T4 Ligase, New England Labs
- 1 µl T4 Ligase (10.000 U)
- 50 ng plasmid
- 3x mol(plasmid) insert
- 2 µl T4 Ligase 10x buffer
- add H2O to reach final volume of 20 µl
- incubation at 22°C for 1 h
- storing at 16 °C for 40 min
Biobrick Standard
[http://parts2.mit.edu/wiki/index.php/Standard_Assembly Standard BioBrick assembly]
Close
Read more
At Woehlke's S1-Lab !!!
- Thaw competent cells on Ice
- Add DNA, pipette gently to mix
- Let sit for 30 minutes on ice
- Incubate cells for 45 seconds at 42°C
- Incubate cells on ice for 2 min
- Add 1 ml LB0
- Incubate for 1 hour at 37oC on shaker
- Spread 100-300 μl onto a plate made with appropriate antibiotic.
- Grow overnight at 37 °C.
- Save the rest of the transformants in liquid culture at 4 °C
modified from [http://openwetware.org/wiki/Transforming_chemically_competent_cells open wetware]
Close
Read more
Agarose Gels
usual volume needed: 80 ml
[http://www.promega.com/enotes/faqspeak/fq0065.htm Optimum resolution according to NEB]
[http://www.cwcboe.org/19992051412432550/lib/19992051412432550/Molecular%20Biology/Gel%20electrophoresis/Lab_manual_8_gel_elect.pdf further information on optimizing gel electrophoresis, e.g. recommanded voltage per cm2 gel]
stain
- SybrGold ([http://www.invitrogen.com/site/us/en/home/References/Molecular-Probes-The-Handbook/Nucleic-Acid-Detection-and-Genomics-Technology/Nucleic-Acid-Detection-and-Quantitation-in-Electrophoretic-Gels-and-Capillaries.html invitrogen])
- Cover Gel with 1x TAE
- Add SybrGold to a 1:10000 dilution
- cover with aluminium foil (light sensitive)
- shake&incubate 20 min (for 2% Agarose Gels at least 45 min!)
standards
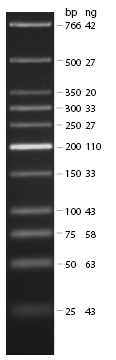
| 
| 
|
low molecular weight (NEB)
| 1 kb standard (NEB)
| 2-log standard (NEB)
|
Polyacrylamide Gels
Preparation of Gels
Recipe for denaturing gels:
| Gel type
| 1 big gel
| 2 big gels
| 1 small gel
| 2 small gels
|
| Urea
| 28.8 g
| 57.6 g
| x
| x
|
| Acrylamide 40%
| 22.5 ml
| 45 ml
| x
| x
|
| Buffer 10x
| 6 ml
| 12 ml
| x
| x
|
| End volume (reach by adding water)
| 60 ml
| 120 ml
| x
| x
|
| APS
| 600 µl
| 1200 µl
| x
| x
|
| TEMED
| 60 µl
| 120 µl
| x
| x
|
- Dissolve Urea in Acrylamide-buffer mixture (use Ultrasound bath), this may take more than an hour!
- Tighten the Gel chamber
- add water to desired end volume
- Add APS, then TEMED, mix
- Pipette mixture into gel chamber
- Add desired comb
- let gel polymerize overnight; add buffer in the evening
Running of Gels
mix samples 1:1 with formamide loading dye (stored @ -20°C)
carefully remove comb
blow air into pockets with a 50 µl syringe
fill samples into pockets
run the gel
(usually about 200 V)
Close
Close
In vivo Measurement
Read more
Bacterial Cell Growth
Bacteria from over night cultures were diluted 1:50 into 20 ml culture in LBamp and incubated at 37 °C. Upon OD600 of 0.7-0.8 the cultures were induced with 0.4% Arabinose and 0.4% Arabinose + 1mM IPTG, respectively. Subsequently Cultures were incubated at 25°C for at least 12 h.
Fluorescence Measurement
Cell samples for the fluorescence measurement were diluted to OD600=0.03 and analyzed in a JASCO fluorimeter. eGFP excitation wavelength was set to 501 nm and mCherry fluorescence was measured with an excitation at 587 nm. Standard parameters for the fluorimeter included scanning speed of 100 nm/ min and data points every 0.2 nm as well as medium detector sensivity. The cuvette holder was temperated to 25 °C.
The resulting spectra were corrected for instrumental wavelength dependencies and quantum yield of the fluorescent proteins. A pure LBamp spectrum was subtracted and the corrected spectra were normalized using eGFP fluorescence as reference.
Close
In vitro Translation
Read more
The [http://www.promega.com/catalog/catalogproducts.aspx?categoryname=productleaf_335&ckt=1 Promega Kit] is used according to the provided protocols. Further Information about this Kit can be found in the [http://partsregistry.org/Cell-free_chassis/Commercial_E._coli_S30 Parts Registry].
Fluorescence kinetics are recorded for at least 3 hours, settings are applied as in the in vitro measurement.
Close
In vitro Transcription
Read more
Buffers
Three different buffers were used for in vitro transcription experiments:
- For Epicentre E. coli RNA Polymerase the recommended buffer was used
- For T7 RNA Polymerase experiments two buffers were tested:
- T7 RPO buffer as recommended and used by members of the Simmel group
- T7 RPO "paper buffer", as used in the paper of XXX
Of all buffers 2x concentrated stocks were prepared. Malachite green was usually added to the buffer stocks.
|
| E. coli RPO buffer
| T7 RPO buffer
| T7 RPO buffer "paper"
|
| Tris
| 40 mM
| 40 mM
| 40 mM
|
| pH
| 7.5
| 7.1
| 7.9
|
| MgCl2
| 10 mM
| 40 mM
| 6 mM
|
| KCl
| 150 mM
| /
| 100 mM
|
| Triton X-100
| 0.01%
| /
| /
|
Sample Preparation
Different concentrations were tested for malachite green and DNA templates. Components of a standard experiment are listed in the table below.
|
| E. coli RPO buffer
| T7 RPO buffer
| T7 RPO buffer "paper"
|
| buffer
| 1x
| 1x
| 1x
|
| DTT
| 10 mM
| 10 mM
| 10 mM
|
| Malachite green
| 5-10 µM
| 5-10 µM
| 5-10 µM
|
| NTP's
| 1 mM
| 4 mM
| 0.8 mM
|
| RPO
| 2 U
| 125 U
| 125 U
|
In each run up to 4 samples are measured simultaneously. Components that are the same in each of the 4 samples (buffer, DTT, NTPs, RPO, Water) are prepared as a 4.1x MasterMix in a loBind tube and split to the 4 cuvettes. Final Volume of each sample is 100 µl.
Cary Eclipse
For fluorescence measurements, a Cary Eclipse Spectrofluorimeter with a Multicellholder (4 cells) is used. Kinetics are recorded at 37° C. Excitation wavelength is 630 nm, emission is followed at 650 nm and 655 nm. After each kinetics measurement, spectra are to be recorded.
Close
References
[1] http://www.promega.com/catalog/catalogproducts.aspx?categoryname=productleaf_335&ckt=1
[2] Zubay, G. (1980) Meth. Enzymol. 65, 856–77, Zubay, G. (1973) Ann. Rev. Genet. 7, 267–87.
|
 "
"










































































































































{kind=link}
{kind=link}
{kind=link}
{kind=link}