|
|
Experiment Design
We designed different experiment set-ups with different complexity to test RNA signal/switch pairs based on our concept.
Our initial idea to prove our concept of antitermination was to use fluorescent proteins as reporters. This approach gives the opportunity to measure the termination and antitermination efficiency of our designed BioBricks in vivo as well as in vitro, the latter using a translation kit based on E. coli lysate. Later on, we decided to developed an experiment, that relies only on transcription. In this, we used a fluorescent dye, malachite green, that binds a specific RNA aptamer and thus makes it possible to detect transcription activity.
In vivo Measurements
In vivo measurements have the highest complexity compared to any other experiment set-up. Our system has to deal with several circumstances a cellular environment comes with, such as interaction with other RNAs, degradation by RNases or unspecific interactions. Nevertheless, the measurements are essential, as our switches should finally work inside cells to fulfill our vision of an intracellular logic network.
Read more
Design
For the measurements in vivo we decided to use an expression cassette consisting of Green Fluorescent Protein (GFP) coding sequence upstream of the switch and another fluorescent protein coding sequence downstream of it. Both protein coding sequence carry the same ribosome binding site, therefore, the GFP fluorescence can be used as internal control in measurements. Since the spectra should not overlap and to avoid FRET as well as an pure overlap of the spectra, we settled on the usage of red fluorescent protein variants, namely mRFP1 in the first try. While the GFP fluorescence is used to normalize the measurements, the RFP fluorescence is used to detect termination/antitermination.
Upon binding of the signal, the stem loop of the switch would resolve leading to red fluorescence. The GFP fluorescence as internal control carries the advantage that errors in the measurement set can be detected easily. Lack of arabinose or promoter insensitivity can be recognized as well as problems with the fluorescence measurement itself. Plus, we have a way to normalize our measurements and compare different preparations in relation to each other.
Construction and Cloning
Our measuring plasmid is based on the BioBrick pSB1A10, A1, distribution 2010. Unfortunately after two months of cloning we had to recognize that the plasmid in use did not work (see also Biobrick validation--> link). So after the first unsuccessful attempts we decided to reclone the system, substituing RFP to mCherry, a dsRED derivative with a spectra in the far red, and adding arabinose inducible promoters in front of both fluorescent proteins.
To control the expression of the switch, the particular DNA sequence itself is under the control of an IPTG dependent promoter. In the future we want our networks to be able to respond to a variety of external signals like small metabolites, ions or whatever can be found in the parts registry. For a start we went with an established and well-working system like the lac-operon. ---> PLASMID MAP
So upon induction with arabinose a rise of GFP expression can be seen. To monitor changes in gene expression we put our E. coli cells in a fluorimeter and measured fluorescent. It worked quite nicely with living cells in a fluorimeter, the only thing to avoid is too much scattering: the cell density should not exceed an OD600 of ???. Continious stirring and a set temparature at 37°C allowed measuring over severall hours. Cell density was checked in between.
Measurement
For switch evaluation, IPTG was added to the cells after about two hours after arabinose induction (baseline). ??? Stimmt das?? A rise of RFP/mCherry emission should be visible in case of a working switch.
For evaluation of the measuring plasmid itself we incorporated a positive control in every measurement. A random sequence in between GFP and RFP/mCherry was chosen in a corresponding length. An increase in both GFP and mCherry was detectable in the positive control and in the same amount after quantum yield correction, proving that the measuring plasmid is working nicely.
As a negative control we measured the same plasmids as for every switch but without signal. Since our switches are effective terminators if no signal is bound, transcription can not occur and no RFP/mCherry is produced.
When measuring the termination of our BioBricks and the antitermination by their corresponding signal-RNA, we should be able to observe an increasing RFP emission compared to the GFP emission upon induced signal-RNA production in the cells/in the kit:
With these measurements, it should also be possible to observe differences in efficiency of termination as well as antitermination between our designed switches.
Close
In vitro Translation
In vitro measurements with E. coli lysate make the fluorescence signals independent of cell growth and physical or biological factors, e.g. cell density or growth stadium.
Read more
Design
In this assay we used the same constructs as engineered for the in vivo studies.
Measurements
We used the cell-free E. coli S30 extract system for circular DNA provided by promega[1], which is prepared by modifications of the Method Zubay et al.[2] described. The characterization of the kit can be obtained from the [http://partsregistry.org/Cell-free_chassis/Commercial_E._coli_S30 Parts Registry].
We perform our experiments at 37°C with an amount of approxim. 1 ug plasmid in a reaction volume of 50 uL. Measurements are carried out in a jasco fluorolog, where the fluorescent proteins RFP and GFP are excited and fluorescence is detected. A general problem occuring in the experiments was the low capacity of the kit. The signal intensity is very low, which made it difficult to observe any signal intensity alterations.
Close
In vitro Transcription
An experiment, in which we detect In vitro transcription, offers an elegant way for a fast and easy prove of principle, since our switch is RNA-based and the whole mechanism takes place on trancriptional level. Most side effects occuring in a complex environment given in a cell or a cell lysate do not arise here.
Read more
If measureable effects with our basic concept can be seen in vitro we can use the so gained data to optimize the system in vivo. Since we are working on a totally new principle of trancriptional control, we used this approach for easy variation of different variables like the length of the core unit and the switch to signal ratio.
To study the switches on the transcriptional level gives the advantage, that we would have less interferences and possible artefacts. Also, we are not sure how cellular mechanisms like degradation of RNases or interacting factors as well as molecular crowding influence our systems.
Working with in vitro systems also has the advantage that an input is not needed anymore and the output can also be generated easily. We used two readouts with two different transcription systems to check and investigate our devices: First, we used an malachitegreen-binding aptamer for an fluorescent output (which will be described in detail later) and second, we simply put our reaction educts on an denaturing acrylamide-gel to check for RNA varying in length. As for two different transcription systems we used on the one hand E. coli-RNA Polymerase (RPO) based transcription since the aim is to apply our system in vivo and on the other hand T7 based transcription which is well established through literature.
T7 RNA polymerase
The T7 RNA polymerase is known for satisfying RNA yields together with easy handling. In our approach we had PCR amplified, double stranded switches with an malachitegreen binding aptamer following after the switch (133 bp, see section below) and a single stranded signal with about 30 bp length.
For in vitro expression the T7 RNA Polymerase requires a double stranded promotor region at the beginning of the DNA template but is otherwise capable of handling single stranded DNA, so a sense strain corresponding to the T7 promoter region was added. Transcription is more effective with double stranded DNA as template. Since we ordered the signal sequences we tested we chose the cheaper way in the beginning by using single stranded signals with corresponding sense T7 pieces and switched to double stranded constructs after narrowing down the most promising switch/signal pairs.
E. coli RNA polymerase
In comparison to the T7 RNA Polymerase the E. coli RNA Polymerase requires slightly more sophisticated proceedings when it comes to the design of switches and handling of the enzyme. The biggest in our case was to store it properly since the only -80°C fridge was in another building.
E. coli RPO was ordered saturated with σ70-factor. The switch consists of an ???-promoter, the switch itself and a ???.
Denaturing Polyacrylamide gel electrophoresis
We also used Polyacrylamide gel electrophoresis (PAGE) for evaluation of termination efficiency of our basic units. Gels containing 15 % acrylamide and 6 M urea were used for separation of 90 (terminated by switch) and 133 bp (continous reading) RNAs.
Polyacrylamide gels seperate RNA and DNA according to their size in an electric field. Since the negative charge equals the size of nucelotides in the RNA/DNA, the number of base pairs can be compared between two samples often with one base pair resolution. Since RNA forms three-dimensional structures, the samples are preheated and run in 6 M urea. The polyacrylamide gel is stained in SybrGold afterwards which binds to both single and double stranded DNA and RNA.
Denaturing PAGE is a simple yet elegant way to check for transcription efficiency and termination rates. Since it is a very direct way and it provides a simple yet clear readout, we used it as another method beside the more sophisticated malachitegreen binding assay to evaluate and characterize our switch.
Malachite green assay
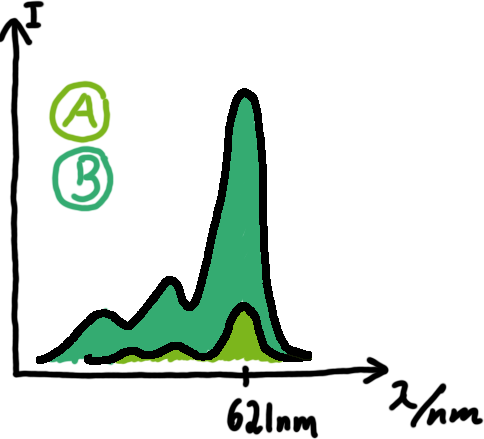 Emission spectra of malachite green; A: without signal-RNA, B: with signal-RNA Malachite-green is a dye with a negligible fluorescence in solution but undergoes a dramatic increase if bound by a RNA -aptamer. Upon binding to the aptamer, the fluorescence of malachite-green increases about 3000 times making it an exceptionel good marker. Since the binding is very specific, transcription in dependence of a signal can be monitored by measuring the fluorescence of malachite-green over time if the aptamer is located behind the switch. Transcription of the aptamer will only take place after anti-termination by a signal. An increase should be visible over time.
For the T7-based measurements we ordered single stranded signals for a first attempt and added matching single strands complementary to the T7 promoter region. The switch was amplified using PCR and consisted of the following elements: Primer-binding site - T7 promotor - switch - malachitegreen binding aptamer. Upon binding of a correct signal to the switch, the stem loop dissolves and transcription is possible.
OLD: A second possibility to measure parameters of our switches we came up with, was the idea to investigate our system on the transcriptional level only. Therefore, we decided to use malachite green as reporter. Malachite green in a fluorescent dye, whose emission increasing dramaticly (about 3000 times) upon binding of a specific RNA-aptamer. The RNA-aptamer
---concept to be described, as well as literature---
<ref>refs</ref>
We made constructs comprising of a sigma(70)-binding promoter followed by a short nonsense sequence, the switches and the aptamer sequence.
Also we made constructs, where the transcription of the signal-RNA is under the control of a sigma(70) promoter. These two linear DNA-constructs, together with the e.coli RNA-polymerase and the right buffer conditions should represent an easy-to-handle measurement kit on the transcriptional level.
Close
References
[1] http://www.promega.com/catalog/catalogproducts.aspx?categoryname=productleaf_335&ckt=1
[2] Zubay, G. (1980) Meth. Enzymol. 65, 856–77, Zubay, G. (1973) Ann. Rev. Genet. 7, 267–87.
Experimental Results
In vivo Measurements
In vivo
In vitro Measurements
Read more
Protocols
Molecular Biology
PCR
Read more
Digestion
Read more
Ligation
In vivo Measurement
Read more
So haben wir in vivo gemessen
Close
In vitro Translation
Read more
So haben wir in vitro gemessen
Close
In vitro Transcription
Read more
So haben wir in vivo gemessen
Close
Lab Book
Explanations
In the following we present an overview regarding our work in the lab. For easier understanding we summarized the work of each week using colored boxes. To get more information about the work and results of a specific week, just click on the according week number. To get a better overview we used the following color code for the boxes:
| This box represents general cloning that were requirde for several measurements. See the protocol section for further details.
|
| The blue box indicates in vivo measurements which are described here.
|
| The yellow box represents measurements done with an in vitro translation kit and is described in more details here.
|
| The green box indicates in vitro measurements relying on in vitro transcription and malachite green measurements. Details can be found here.
|
Chronological Lab Book
Week01
Constrcuts for in vivo measurments
08.04.2010
Flo & Philipp
PCR
- samples:
- R0011_His
- R0011_Trp
- Control
- protocol: protocols
- templates: purified PCR products from 5.2.2010
- primer G1004/1005
- polymerase: Taq
- programm: igempcr
Purification of PCR products with QIAquick PCR purification Kit
- protocol followed. exceptions: DNA-binding/unbinding with 3min 6000rpm followed by 60sec full speed
2% Agarose Gel
09.04.2010
Philipp & Flo
Gel of PCR products from 08.04.2010
- loaded: 10 uL sample+2 uL 6x GLD, 4/2 uL LMW standard
- 110 V, 90min
- stained with Sybrgold, 20min, 1:10.000 dilution in TAE
- Standard - Control - R0011_His - R0011_Trp - Standard(=low molecular weight (see Protocols#Lab_Protocols))
File:100409.JPG
Close
Week02
Improved Constructs... Testing
15.04.2010
Philipp & Flo
[http://web.e14.physik.tu-muenchen.de/igem/index.php/Protocols#PCR PCR] of B0014 and R0011
16.04.2010
Philipp & Flo
- Concentrations measured with nanodrop:
| B0014
| 2.5 ng/uL
|
R0011
| 27.5 ng/uL
|
--> worked for R0011, not for B0014
- PCR of B0014
- Purification with the Zymo Kit, Elution in 20 uL H2O
- Concentration measured with nanodrop, 17.5 ng/uL --> worked
template
| restriction enzymes (biobrick assembly)
|
B0014 (from Christoph, verified PCR products, 21 ng/uL)
| EcoRI, PstI
|
R0011 (from PCR [15042010], 27.5 ng/uL
| SpeI
|
HisSig (1:100 dilution)
| XbaI
|
TrpSig (1:100 dilution)
| XbaI
|
| psB1K3 (with RFP insert, from HiWiPhilipp, 81 ng/uL)
| EcoRI, PstI
|
5 uL template used for each setup. protocol followed
- Gel for purification of the cleaved plasmid
- 2% Agarose in 1x TAE
- 120 V, 90 min
- stained with SybrGold
- digestion, digestion, 1 kb ladder
- Digestion worked (partly). band at 2000 bp (backbone) cut
File:100416.png
- Purification of DNA from Gel
- Ligation of HisSig/TrpSig with R0011in 2 reactions
| used Volume
| approx. concentration*
|
HisSig
| 6 uL
| 7 ng/uL
|
TrpSig
| 6 uL
| 5 ng/uL
|
R0011
| 3 uL
| 6 ng/uL
|
* approximated from the amount used in the digestion before
Close
Week03
Measurements
19.04.2010
- PCR of R0011-TrpSig and R0011-HisSig
- Purification with the Zymo Kit, Elution in 30 uL H2O
- Concentration measured with nanodrop: c(R0011-TrpSig)=20 ng/µL, c(R0011-HisSig)=12.5 ng/µl --> worked
- Gel for analysis of ligation and PCR
- 2% Agarose in 1x TAE
- 110 V, 90 min
- stained with SybrGold 1:10000 20 min
- pure R0011 PCR product used as control
File:GEL 20100419beschriftet.png
LMW
| 4 µl
|
R0011-TrpSig
| 5 µL
|
R0011-HisSig
| 5 µL
|
R0011
| 5 µL
|
Samples seem to have run further than the buffer/dye-Front! But: Ligation Products show bands at shorter lengths than R0011 alone --> Ligation didn't work ?!?
- Ligation of HisSig/TrpSig with R0011in 2 reactions
| used Volume
| concentration
|
pSB1K3
| 5 uL
| 10 ng/µL (nanodrop)
|
B0014
| 3 uL
| 5 ng/µL approx.*
|
* approximated from the amount used in the digestion before
20.04.2010
- Gel for analysis of ligation and PCR (repeat of yesterday's gel)
- 2% Agarose in 1x TAE
- 130 V, 75 min
- stained with SybrGold 1:10000 60 min
- pure R0011 PCR product used as control
- Excision and purification of marked bands at 200bp using QIA Kit, elution in 30 µl H2O
File:Gel100420marked.png
- PCR of excised and purified bands of R0011-TrpSig and R0011-HisSig
- complete samples (30 µl) used as templates
- Purification with the Zymo Kit, Elution in 30 uL H2O
- Concentrations of PCR-products: 0.5-1 ng/µl --> Gel excision or PCR didn't work
- Transformation (Woehlke-Lab)
- 8 µl of ligation product pSB1K3-B0014 to 50 µl XL-10 competent cells
- 200 µl plated on a Kanamycin-containing Plate
- remaining 800 µl stored @4°C in S1-lab
21.04.2010
- Gel for analysis of ligation and PCR (repeat of yesterday's gel)
- 2% Agarose in 1x TAE
- 110 V, 90 min
- stained with SybrGold 1:10000 80 min
- pure R0011 PCR product used as control
- Excision and purification of marked bands at 200bp using Zymo 5 Kit, elution in 20 µl H2O
File:100421beschriftet.gif
- PCR of excised and purified bands of R0011-TrpSig and R0011-HisSig
- complete samples (20 µl) used as templates
- Purification with the Zymo Kit, Elution in 25 uL H2O
- Concentrations of PCR-products:
- R0011-TrpSig: 22.5 ng/µl
- R0011-HisSig: 9.5 ng/µl
--> worked!!!!!
- 'Colony PCR
- 7Colonies picked and resuspended in 20 µl LB+Kana (each)
- PCR of 2 µl of each sample, 2 µl as negative control (Program: ColonyPCR, modified)
- 15 µl of each sample mixed with 3 µl GLPn and loaded to Gel
File:100421colony.png
- Overnight cultures:
- remaining 18 µl of samples 1, 3, 6, and 7 added to 5 ml LB + kanamycin
- 37°C on Shaker
22.04.2010
- Gel for purification of PCR products R0011-TrpSig and R0011-HisSig (yesterday's result)
- 2% Agarose in 1x TAE
- 110 V, 90 min
- stained with SybrGold 1:10000 30 min
- pure R0011 PCR product used as control
- Excision and purification of marked bands at 200bp using Zymo 5 Kit, elution in 20 µl H2O
File:100422beschriftet.png
- Miniprep
- Result: about 4 µg Plasmid
23.04.2010
template
| template volume
| restriction enzymes
| Buffer
|
HisSig (1:100 dilution)
| 5 µl
| EcoRI, SpeI
| NEB4
|
TrpSig (1:100 dilution)
| 5 µl
| EcoRI, SpeI
| NEB4
|
| psB1K3-B0014 from Miniprep (No 7, 35 ng/µl)
| 5 µl
| EcoRI, XbaI
| NEB4
|
Incubated 90 min @ 37°C
- Gel for purification of the cleaved plasmid
- 2% Agarose in 1x TAE (leftover from yesterday)
- 140 V, 90 min
- stained with SybrGold 40 min
- 4 µl 1 kb ladder, 10 µl purified digestion + 2 µl GLPn, 10 µl purified digestion + 2 µl GLPn
- Digestion worked (partly). band at 2400 bp cut out
File:100423beschriftet.png
- Purification of DNA from Gel
- A260/A230 and A260/A280 values were strange (see labbook)
Close
Week04
Cloning Measurements Measurements Measurements
Week05
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week06
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week07
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week08
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week09
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week10
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week11
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week12
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week13
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week14
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week15
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
Week16
Cloning Measurements Measurements Measurements
This is what we did in week xxx.
Close
|
 "
"














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}