The addition of the catechol to a suspension of cells expressing catechol 2,3-dioxygenase will result in the production of 2-HMS.
In our second experiment we wanted to measure the absorbance of 2-hydroxymuconate semialdehyde over time. We grew a 5 mL culture of our engineered E. coli cells in M9 media overnight. 1 mL of the cell solution was removed for analysis. Catechol was added to a final concentration of 100mM in the cell suspension. Immediately the formation of 2-HMS was tracked. The formation of 2-HMS can easily be tracked, as it absorbs light at 375 nm.
Catechol 2,3-dioxygenase contains an iron molecule in its active site. Our hypothesis was that the iron molecule is oxidized after converting a single catechol molecule to 2-HMS; rendering the catechol 2,3-dioxygenase inactive. We also want to test how catechol 2,3-dioxygenase would behave in vitro compared to in vivo.
To test this hypothesis we grew E. coli containing BBa_K118021 in 500 mL of LB media with ampicillin. E.coli containing the pUC19 plasmid were also grown in 500 mL of LB media with ampicillin to act as a negative control throughout the course of the experiment. The cells were grown to a final optical density of 4.55 AU (measured at 600 nm). The cells were first spun down at 3800 rcf for 5 minutes. The supernatant was decanted and the cells were re-suspended in 40 mL M9 minimal media and incubated with 10 mg of lysozyme for 10 minutes. This cell extract was spun down at 10000 rcf for 30 minutes. The supernatant was taken off and spun at 30000 rcf (S30) for 1 hour. The supernatant of the S30 samples was divided. Half the samples were flash frozen in liquid nitrogen and stored at -80oC. The second half was spun at 100000 rcf for 45 minutes.
In order to measure the 2-HMS production we needed to determine the concentration of protein per volume of cell extract. To determine the total protein concentration of the S30 and S100 extracts a Bradford assay was conducted using a standard curve of BSA. Concentrations of the negative controls (Escherichia coli DH5α hosting the pUC19 plasmid) for each of the S30 and S100 samples were also determined. Absorbance readings were taken at a 595 nm wavelength and concentration reported in µg/mL.
| Sample | Concentration
|
| S30 | 779µg/mL
|
| S30 Control | 104.5µg/mL
|
| S100 | 519.3µg/mL
|
| S100 Control | 100.1µg/mL
|
Protein mass of S30 and S100 (and S30 Control/S100 Control) extracts were normalized to between 2µg and 10µg in 1mL of either 20mM Tris pH 8.0 or double distilled water. Catechol was added to normalized cell extract at a final concentration of 0.05 mM. The subsequent production of 2-HMS was observed as a function of time by recording absorbance at 375 nm over the ten minute period immediately following the addition of catechol.
Results
Figure 4. displays that xylE can degrade catechol in vitro producing 2-HMS. We also observe that when roughly double the amount of S30 extract is added to an excess catechol solution roughly double 2-HMS is produced. This suggests that the enzyme is the limiting component in catechol degradation by xylE. This result corresponds with our hypothesis that the xylE active site iron is reduced, rendering our enzyme inactive. The samples in Figure 4. were carried out in double distilled water. However, dublicates of the readings were measured in a buffer (20mM Tris pH 8.0).We did not observe a significant difference in 2-HMS production in samples with Tris buffer, vs. unbuffered samples containing double distilled water.

|
|
Figure 3. The production of 2-hydroxymuconate semialdehyde by different concentrations of S30 extract. Green and red lines are 2µg and 4µg respectively of S30 cell extract from cells expressing catechol 2,3-dioxygenase. The blue line is 4µg of control S30 cell extract.
|
|---|
Conclusion
Justin and Adam to fill in!
Compartmentalization Parts
One of the sub-projects for the bioremediation of the tailings ponds is to create synthetic microcompartments that we can then use to isolate various pathways within an Escherichia coli cell. To do this we need to have a microcompartment as well as a means to characterize the compartment so that the system can be optimized. Here are the experiments we have performed so far towards the characterization of the microcomparments.
Placement of Oligoarginine Tail on Proteins
Characterized Parts
BBa_K249004
BBa_K249005
BBa_K331030
BBa_K331031
Hypothesis
The placement of an oligoarginine sequence at the N-terminus of a protein will destabilize the protein in vivo.
Introduction
The long term goal of our team is to utilize an oligoarginine tail to specifically target enzymes into a microcompartment composed of modified lumazine synthase subunits. While conducting background research on the project, we came upon data originally reported by Bachmair et al.1 suggesting that the identity of the amino acid at the N-terminus of a protein is related to its half-life, and mostly notably, that arginine residues at the are destabilizing. This data suggests that by placing an arginine at the N-terminus of a protein to be targeted into a lumazine synthase microcompartment would cause degradation of our protein before it can be moved into the microcompartment.
We chose to investigate the how the placement of an oligoarginine sequence affects the stability of the protein to which it is fused.
Method
In order to further characterize the C-terminal and N-terminal oligoarginine tag (BioBricks BBa_K249005 and BBa_K249004 respectively) and investigate the effect their placement on protein stability, yellow fluorescent proteins (YFP) with the oligoarginine fused to either the C-terminus (BBa_K331023) or N-terminus (BBa_K331022) (and preceded by a ribosomal binding site – BBa_B0034) were synthesized. We used our Red/White 3-Antibiotic assembly method to add a tetracycline repressible promoter (BBa_R0040) for constitutive expression of the fusion protein. This addition generated BioBricks BBa_K331031 and BBa_K331030 for the C-terminal tagged and N-terminal tagged YFP respectively.
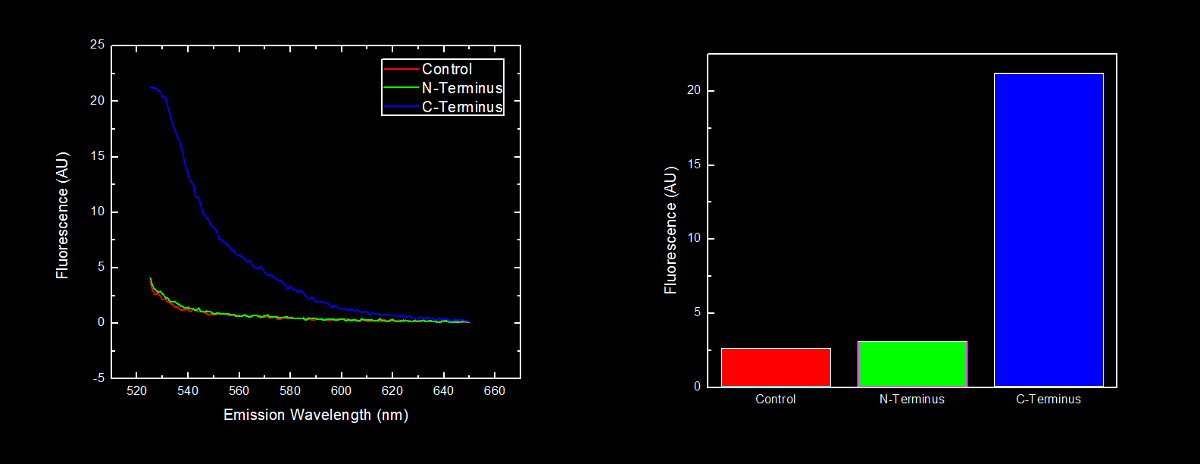
The BioBrick containing plasmid was transformed into Escherichia coli DH5α cells. These cells were grown to an OD600 of approximately 0.7, and diluted 1:10 with MilliQ H2O immediately prior to analysis by fluorescent spectroscopy.
This dilution of cells was excited at 517 nm, and the emission spectra was read from 522 nm to 650 nm. Fluorescence at 524 nm (emission maxima of YFP) of control cells (Escherichia coli DH5α), N-terminal tagged, and C-terminal tagged YFP were compared.
Results
N-terminal tagged YFP did not have substantially more fluorescence than control cells. Cells expressing C-terminal tagged YFP had ten times more fluorescence than control cells and cells expressing N-terminal tagged YFP.
Conclusion
Our results showing that the placement of arginine residues at the N-terminus of our YFP results in no observable fluorescence over control cells are consistent with the general N-terminal rule reported by Bachmair et al. Assuming that transcription of this K331030 and K331031 are equivalent, these data suggest that the N-terminal oligoarginine is reducing the half-life of the protein to which it is fused, ie YFP.
Reference
1Bachmair A., Finley D., Varshavsky A. (1986), In Vivo Half-Life of a Protein Is a Function of Its Amino-Terminal Residue. Science 234. 4773 179-186.
Characterization of C-terminal Oligo Arginine Cyan Fluorescent Protein
Characterized Parts
BBa_K331033
Hypothesis
Light will be observed at 476 nm when cells containing BBa_K331033 are illuminated with light at 439 nm as a result of the excitation and subsequent fluorescence of Cyan Fluorescent Protein (CFP).
Introduction
Prior to introducing a oligoarginine tail onto the catechol-2,3-dioxygenase protein with the intention of directing the fusion protein into a lumazine synthase microcompartment, there must be some confirmation that oligoarginine tail is indeed causing the protein to which it is attached to localize within the microcompartment. We chose to utilize a technique known as fluorescent resonance energy transfer1 (FRET) to verify localization. In FRET, when a dye pair comes into close proximity to one another, one dye (the donor) will transfer some of its energy to the second dye (the acceptor). This will cause the acceptor to fluoresce without direct excitation. The dye pair we have chose is CFP as the donor and yellow fluorescent protein (YFP) as the acceptor.
Here we investigate the ability of BBa_K331033 to produce CFP and emit light at its characteristic emission wavelength.
Method
In order to characterize the tetracycline repressible CFP (BioBrick BBa_K331033), cyan fluorescent protein (CFP-BBa_E0020) with an oligoarginine tag fused to the C-terminus (BBa_K331025) (and preceded by a ribosomal binding site – BBa_B0034) was synthesized. We used our Red/White 3-Antibiotic assembly method to add a tetracycline repressible promoter (BBa_R0040) for constitutive expression of the fusion protein. This construction yielded BioBrick BBa_K331033.
The BioBrick containing plasmid was transformed into Escherichia coli DH5α cells. These cells were grown to an OD600 of approximately 0.7 in LB media, and diluted 1:10 with MilliQ H2O immediately prior to analysis by fluorescent spectroscopy.
This dilution of cells was excited at 439 nm, and the emission spectra was read from 444 nm to 650 nm.
Results
Fluorescence at 476 nm was observed. This peak, along with a shoulder occurring at approximately 510 nm is consistent with results obtained by McRae1 et al. in their rapid purification of ECFP.
Conclusion
The BioBrick BBa_K331033 that we constructed generates CFP in the absence of tetracycline, as expected.
Future Directions
We will assembly this into a single biobrick which can differentially express CFP/YFP and lumazine synthase for confirmation (through FRET) that the oligoarginine tail can be utilized to localize proteins within a modified lumazine synthase microcompartment.
Reference
1Förster T. (1948), Zwischenmolekulare Energiewanderung und Fluoreszenz. Annalen der Physik, 437: 55-75.
2McRae S.R., Brown C.L., Bushell G.R. (2005), Rapid Purification of EGFP, EYFP, and ECFP with high yield and purity. Protein Expression and Purification 41. 1 121-127.
Magnetic Nanoparticles Parts
One of the sub-projects for the bioremediation of the tailings ponds is to reduce heavy metals to create magnetic nanoparticles that can then be removed from the pond. To do this we need to be able to produce Mms6 (the iron reducing protein) and show that it can successfully produce the nanoparticles within the Escherichia coli cell. He is the work we have accomplished so far to characterize Mms6.
Characterization of Mms6
Characterized Parts
BBa_K249019
Hypothesis
Induction of Mms6 protein overexpression in Escherichia coli DH5α cells will cause a change in growth.
Introduction
A parallel project we are undertaking in oilsands remediation is in the clean-up of heavy metal contaminants utilizing the Mms6 protein expressed by Magnetiospirilum magneticum1. The Mms6 protein is capable of reducing aqueous iron into magnetite nanoparticles. The magnetite nanoparticles can be removed with much greater ease than aqueous iron.
We chose to investigate how the presence of the Mms6 protein affected Escherichia coli DH5α, its bacterial host cell.
Method
In order to characterize the effect of Mms6 production on Escherichia coli DH5α cells, we utilized BioBrick BBa_K249019, a part submitted by the 2009 Lethbridge iGEM team. BBa_K249019 is the Mms6 coding region (BBa_K249016) with a ribosomal binding side (BBa_B0030) and a double terminator (BBa_B0015) under the control of a lactose inducible promoter ( BBa_R0010).
We inoculated two 500 mL cultures of LB media with Escherichia coli DH5α cells containing BBa_K249019 and grew the cells until they reached approximately 0.6 OD600 units. At this point, expression of the Mms6 protein was induced in one of the 500 mL cultures by the addition of 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG). One milliliter of cells from each sample was removed at one hour intervals and the optical density at 600 nm was recorded and plotted.
Results
The addition of IPTG to cells containing the BBa_K249019 BioBrick caused a slowdown of cell growth after 2 hours, and a subsequent plateau at approximately one-half of the cells density that uninduced cells reached after three hours post-induction.
Conclusion
The slowdown in cell growth as a result of production of Mms6 protein could be attributed to a number of factors.
One potential explanation could be a significant diversion of cellular resources to the production of Mms6 protein. This is unlikely, as subsequent analysis of cell fractions with SDS-PAGE showed no increase in banding at the expected molecular weight.
Another explanation could be that the Mms6 protein is toxic to the cells, and causes them to die. This possibility is being explored in collaboration with the Calgary team by using their troubleshooting kit.
References
1Arakaki A., Masuda F., Amemiya Y., Tanaka T., Matsunaga T. (2010). Control of the morphology and size of magnetite particles with peptides mimicking the Mms6 protein from magnetotactic bacteria. Journal of Colloid and Interface Science. 343:65-60.
 "
"

















