| August
|
| M | T | W | T | F | S | S
|
| | 1
|
| 2 | 3 | 4 | 5 | 6 | 7 | 8
|
| 9 | 10 | 11 | 12 | 13 | 14 | 15
|
| 16 | 17 | 18 | 19 | 20 | 21 | 22
|
| 23 | 24 | 25 | 26 | 27 | 28 | 29
|
| 30 | 31 |
-
|
|
|
|
September
01/09/2010
- gel extraction of amplified cap genes
- 2x DNaseI digestion
- 1st try: 0.5µl DNaseI, 90s, majority <100bp
- 2nd try: 0.4µl DNaseI, 75s, majority <100bp
- gel extraction
- 1st PCR w/o primers
500 ng input from purified DNAse digest
10 ul Phusion HF Buffer
1 ul dNTPs
1.5 ul DMSO
0.5 ul Phusion Polymerase
ad 50 ul H2O
02/09/2010
- PCR of day before: (1kb Plus)
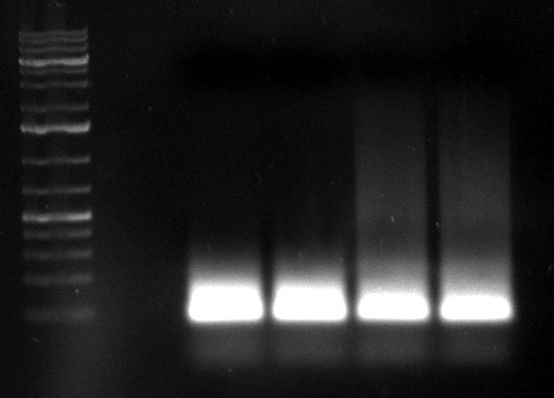
- repetition of both PCRs with cap gene fragments of day before by Dirk
- cap gene DNaseI digestion
| | 3rd try | 4th try
|
| VDNaseI | 0.3µl | 0.2µl
|
| tincubation | 75s | 60s
|
gel image
1kb Plus | 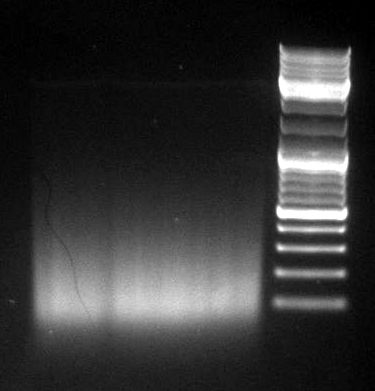 | 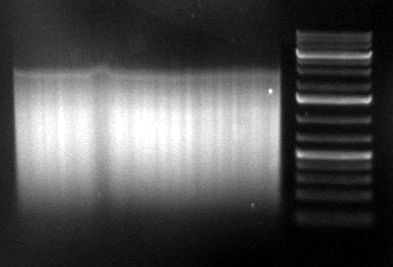
|
majority of
fragments | <100bp | ~1kb
|
- gel extraction
- miselution into collection tube
- nucleotide removal
- c ≈ 37ng/µl in 32µl each ⇒ enough for 3 PCRs each
03/09/2010
- both PCRs with cap fragments of day before
- bands at ~100bp → probably primer binding to small fragments, 1st PCR not working
- no bands at ~2kb → no success
06/09/2010
- cap gene DNaseI digestion
| | 5th try | 6th try
|
| VDNaseI | 0.25µl | 0.25µl
|
| tincubation | 75s | 75s
|
gel image
1kb Plus | 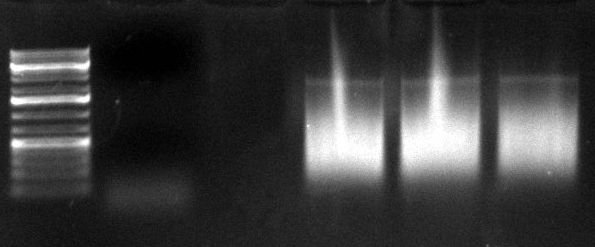 | 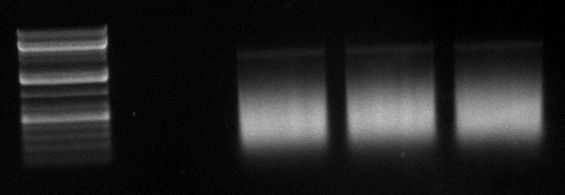
|
majority of
fragments | ~400bp | ~300bp
|
- repetition of whole DNA shuffling by Dirk: success
- assumption: problem at the production of the cap genes → restart of very first PCR the day after
07/09/2010
- amplification of cap genes (1,2,5,6,8,9) out of pBluescript vectors
- 4 aliquots each, only 2 for AAV9
- gel extraction
08/09/2010
- cap gene DNaseI digestion
| | 1 (Thomas) | 4 (Philipp) | 2 (Thomas) | 5 (Philipp)
|
| VDNaseI | 0.3µl | 0.27µl | 0.35µl | 0.45µl
|
| tincubation | 75s | 75s | 75s | 75s
|
gel image
1kb Plus | 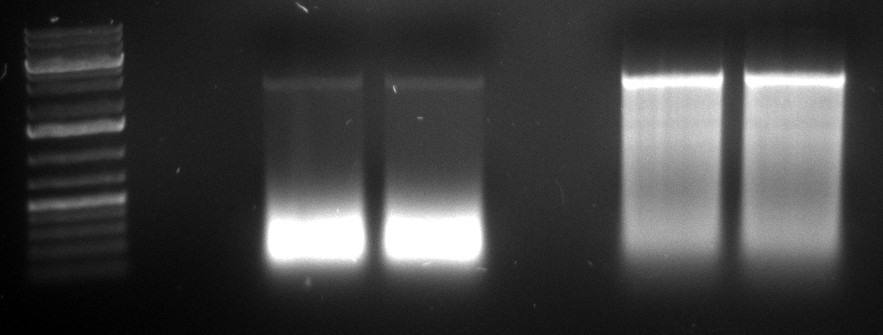 | 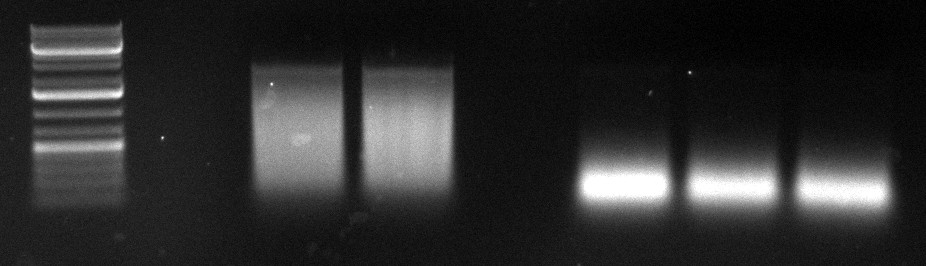
|
majority of
fragments | ~250bp | widespread | ~500bp | <100bp
|
- gel extraction
- 1st and 2nd PCR
- again failed: 1 left, 4 right
- expectation: clear bands at ~2.2kb
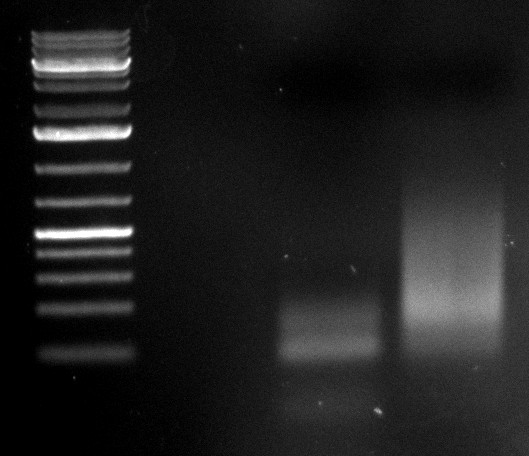
09/09/2010
- 2nd PCR of samples 2 and 5:
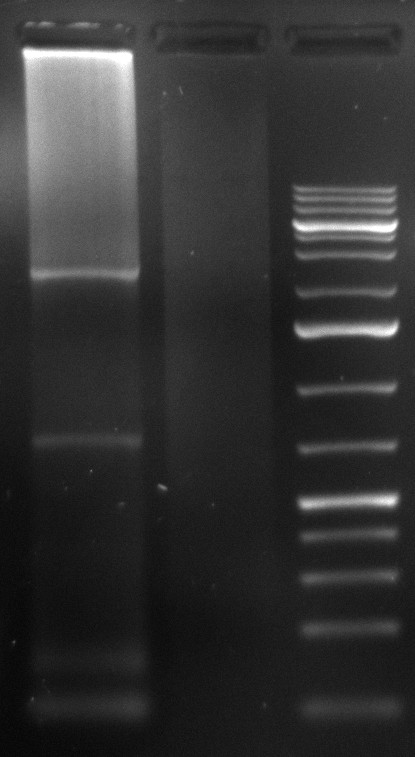
- desired product at 2.2kb visible
- repetition of 3 more PCR samples for preparation
10/09/2010
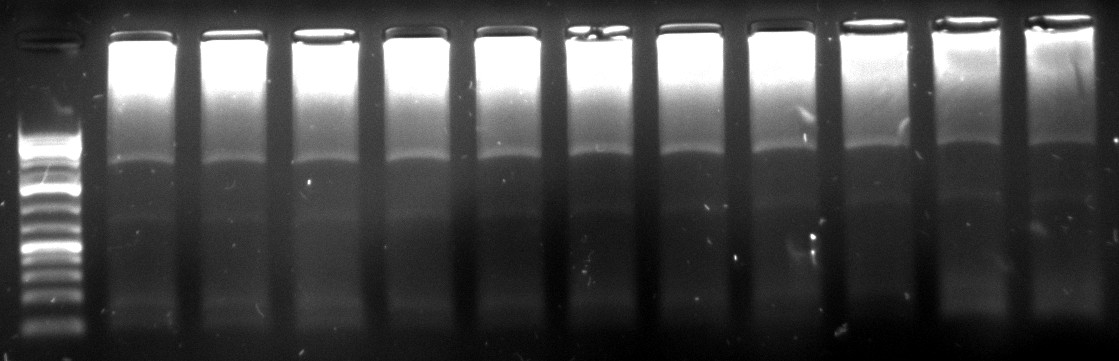
- gel extraction of highest clear band from all lanes
⇑ top
10/09/2010
cap Gene Shuffling (starting over again)
cap PCR
PCR to amplify the cap genes from the stock.
- 5 ul cap plasmid (40 ng/ul stock)
- 1 ul M13For
- 1 ul M13Rev
- 10 ul 5x Hifi buffer
- 1 ul Hifi (Quiagen)
- 32 ul H2O
Cycles (Program name: "shuffle 1st"):
- 95 °C for 5 min
- 94 °C for 15 sec |
- 57°C for 30 sec | 40x
- 68 °C for 3 min |
- 72 °C for 10 min
- 4 °C hold
Gel Extraction:
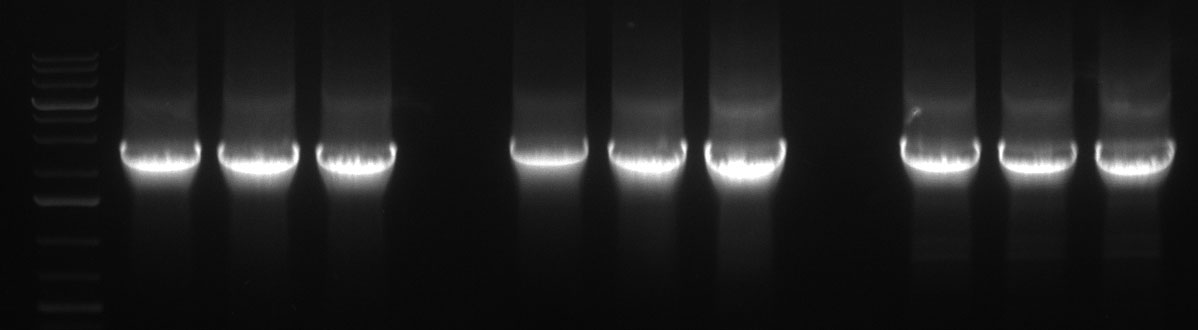 350px
350px
DNAse Digest
cap gene mix (4ug DNA total):
B1/B2 (09/15) !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
AAV m [ng] c [ng/ul] V [ul]
1 350 121.0 2.9
2 700 115.0 6.1
5 1200 93.0 12.9
6 350 105.0 3.3
8 700 172.0 4.1
9 700 186.0 3.8
H2O 20.5
DNAse Digest:
- 4 ug DNA cap gene mix
- 6 ul 10x buffer
- 0.3 ul DNAse I (invitrogen)
Directly after adding of DNAse:
- Flick tube three times
- Start timer 75 sec
- Quick run on the centrifuge (5 sec)
- Incubate on Thermoblock (Eppendorf)
- Add 6 ul 25 mM, vortex briefly
- Inactivate at 75 °C for at least 10 min
Gel Extraction:
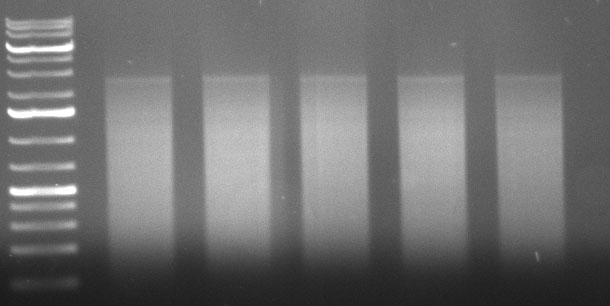
Re-Assembly PCR
500 ng input from purified DNAse digest
10 ul Phusion HF Buffer
1 ul dNTPs
1.5 ul DMSO
0.5 ul Phusion Polymerase
ad 50 ul H2O
Amplification PCR
2 ul Re-Assembly PCR product
1 ul SAFor
1 ul SARev
0.5 ul MgSO4
10 ul 5x Hifi Buffer
33.5 ul H2O
Cycles 2-step PCR protocol (Program name: "shuffle phil", left PCR):
95 °C for 5 min
94 °C for 15 sec |
68°C for 3 min | 40x
72 °C for 10 min
4 °C hold
⇑ top
10/09/2010
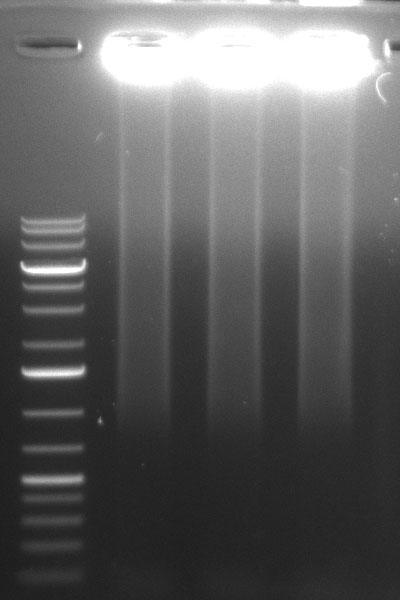
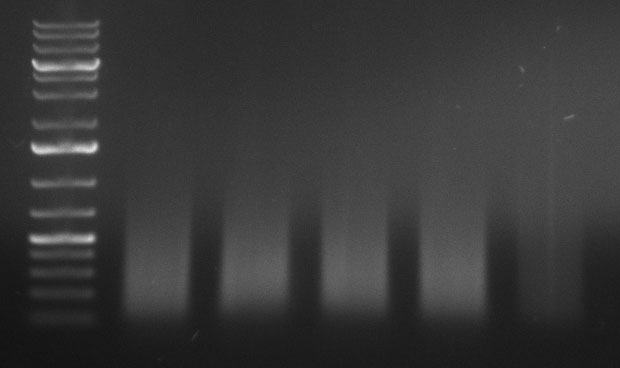
ToDo
ShufflingSchedule
20/09/2010
- PCR-shuffled capsid genes from AAV serotypes 1,2,5,6,8 and 9 were digested with PacI and AscI overnight at 37ͦC according to the following protocol:
- 15 µg capsid genes.
- 7 µL AscI
- 7 µL PacI
- 20 µL 10X NEB buffer 4
- 2 µL 100X
- Up to 200 µL nuclease-free water
- The vector pTR-UF3 was digested with pTR-UF3 using AscI and PacI for 5 hours according to the following protocol:
- 10 µg vector
- 2 µL AscI
- 2 µL PacI
- 5 µL 10X BSA
- 5 µL 10X buffer 4
- Up to 50 µL nuclease-free water
21/09/2010
- The digested capsid fragments and the vector were loaded on a 1% agarose gel and ran for 50 minutes. The ~2.2 KB (capsid genes) and ~5 KB (vector) fragments were then purified from the gel.
- A ligation reaction was carried out according to the following protocol:
- ~3.556 µg insert (Capsid genes)
- ~2.667 µg vector
- 6 µL T4 DNA ligase
- 8 µL T4 DNA ligase buffer
- Up to 80 µL nuclease-free water
- Incubate at 16 ͦC overnight
22/09/2010
- The ligation product from the day before was used to transform electrocompetent cells (Invitrogen), according to the following:
- 600 µL of electrocompetent bacteria were mixed with 62.5 µL DNA from the ligation reaction (about 125 ng per 20 µL bacteria), 20 µL of the mixture were placed in the electoporation cuvette
- Electroporation conditions:
- Recovery of bacteria was done by adding 1 mL LB media to each 20 µL of bacteria that were electroporated. The bacteria were collected in a 200 mL flask and incubated at 37ͦC with shaking at 225 rpm for 1 hour.
The bacteria were then used to inoculate 15 cm petri dishes with ampicillin resistance media. 500 µL were spread on each plate, and 50 plates in total were inoculated. The plates were incubated at 37 ͦC overnight.
23/09/2010
- The plates from the previous day were collected, and 50 colonies were picked and used to inoculate miniprep cultures.
- The colonies from the 50 plates were pooled together and cultured in 700 mL LB for 3 hours, after which a Megaprep (Invitrogen) was done (according to manufacturer's recommendations) to extract the plasmids that contain the shuffled capsid genes and create a library of those shuffled capsids that would be used for the selection of the best capsids, and thus best AAVs.
- A left-over from the transormation from previous day was stored in the fridge and used to inoculate three more 15 cm plates, since the other 50 plates were not suitable for counting the colonies (very crowded!) to estimate the size of the library.
24/09/2010
- 5 flasks (?) of HEK cells were transfected with a total of 75 µg of the library DNA and AAV helper plasmid (1:1) for AAV production, using HBSS. However, the transfection was not successful.
- minipreps of the 50 picked colonies were done, and 10 clones were sent for sequencing. Test digestion with AscI, PacI indicated that the clones picked were positive for the insert (capsid genes).
- Counting of the colonies on the plates from the previous day showed a library size of (...)?
50 more colonies were picked from one of the plates and used to inoculate mini-prep cultures.
25/09/2010
- Analysis of the sequencing results indicated good shuffling of the capsids of two samples, while the 8 others had mainly the sequence of the capsid of AAV5.
- 50 mini-preps of the colonies picked on the previous day were initiated.
27/09/2010
- The 50 samples from the mini-preps from the previous day were test-digested with AscI and PacI, and additionally with NcoI to identify whether AAV5 is present more than the other serotypes. The test digestion with NcoI revealed an AAV5 contamination, so most samples were positive for the AAV5 pattern when digested with NcoI. Sequencing of 10 more samples confirmed this finding.
28/09/2010
- The source of the contamination with AAV5 was identified. The second PCR for the shuffled capsids was repeated.
29/09/2010
- The 2.2 KB band corresponding to cap genes was purified from the gel that was run for the PCR from the previous day. Digestion of the purified cap genes and the pTR-UF3 was done overnight using AscI and PacI according to the same protocol from the previous time.
30/09/2010
- Ligation of the cap genes into the pTR-UF3 vector was carried out accoring to the same protocol from the previous time, the ligation time was 6 hours instead of overnight ligation.
- Transformation of Invitrogen electrocompetent cells was done like the previous time and according to manufacturer's recommendations.
 "
"















{kind=link}