"
"
Team:MIT phage results
From 2010.igem.org
| Line 173: | Line 173: | ||
To facilitate fast debugging, we designed a system that would couple the heterodimerization of GR1 and GR2 to a fluorescent output, via split GFP. | To facilitate fast debugging, we designed a system that would couple the heterodimerization of GR1 and GR2 to a fluorescent output, via split GFP. | ||
<br> | <br> | ||
| - | <img src="https://static.igem.org/mediawiki/2010/a/aa/SplitGFP.gif" width=60%> | + | <a href="https://static.igem.org/mediawiki/2010/a/aa/SplitGFP.gif"><img src="https://static.igem.org/mediawiki/2010/a/aa/SplitGFP.gif" width=60%></a> |
</div> | </div> | ||
Revision as of 22:31, 27 October 2010
hairy cells and polymerizing phage - results |
||||||||
|
Experimental Aims This summer, we pursued three main experimental aims:
Polyphage formation Rakonjac and Model (1998) showed that in the absence of pIII, elongation of the phage proceeds without termination, and the assembling phage stay associated with the cells. We were able to reproduce this phenotype with cells transformed with the hyperphage phagemid. Cells that were transformed with the hyperphage phagemid show a dense mat of many long filaments extending from the cell.
In contrast, when we transformed with hyperphage and our pIII producing plasmid, BBa_K415138, we observed that the polyphage were absent.
Displaying Fusion Proteins In testing for the incorporation of our fusion into the phage coat, we chose to have M13K07 package our fusion instead of hyperphage. Testing with hyperphage would be difficult because the 100-fold reduction in infectivity (Rakonjac and Model, 1998) and the property of being tethered to the membrane introduce complications in the necessary amplification and purification steps. M13K07 and Hyperphage are equivalent in all other aspects, so M13K07 makes an ideal system for testing the ability of our fusions to be incorporated into the phage coat. We transformed cells with our fusions, infected them with the M13K07 helper phage, and purified the resulting amplified phage, then western blotted with HA and Myc antibodies. We did not induce cultures because our fusions were under control of R0065, a very leaky promoter, and we postulated that the basal level of expression would be enough for incorporation. In the western blot, we observed signals from p8-Fos* and p8-GR1. The other linkers may require some tuning of induction or additional troubleshooting before we observe incorporation.
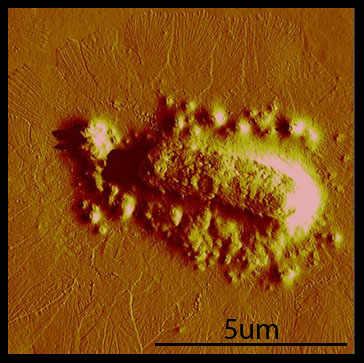
Cross-linking Over the course of the summer, we've found that taking AFM images is difficult and time-consuming. Our original plan was to take AFM images of cross-linking hyperphage, After repeated attempts and little success, we realized that we needed to have a fast way of debugging our system. To facilitate fast debugging, we designed a system that would couple the heterodimerization of GR1 and GR2 to a fluorescent output, via split GFP. 
← Construction
Context →
|