 "
"
Team:Freiburg Bioware/NoteBook/Labjournal/October
From 2010.igem.org
(→Analysis of viral harvest via spectrometer) |
(→Analysis of viral harvest via spectrometer) |
||
| Line 427: | Line 427: | ||
<br /> | <br /> | ||
The plan: Two different viral stocks were prepared: | The plan: Two different viral stocks were prepared: | ||
| - | <br /> | + | <br /><br /> |
| - | Stock 1 with VP2-mVenus-fusion-Capsid loaded with TKGMK | + | Stock 1 with VP2-mVenus-fusion-Capsid loaded with TKGMK (4 wells from a 6 well plate)<br /> |
<ul> | <ul> | ||
<li>R/C 1: 50% pCerulean_VP1up_NLS_mVenus_Vp2/3 P501</li> | <li>R/C 1: 50% pCerulean_VP1up_NLS_mVenus_Vp2/3 P501</li> | ||
| Line 436: | Line 436: | ||
</ul> | </ul> | ||
<br /> | <br /> | ||
| - | Stock 2 "standard" virus | + | Stock 2 "standard" virus (4 wells from a 6 well plate)<br /> |
<ul> | <ul> | ||
<li>R/C 1 P486</li> | <li>R/C 1 P486</li> | ||
| Line 442: | Line 442: | ||
<li>pHelper</li> | <li>pHelper</li> | ||
</ul> | </ul> | ||
| - | These two stocks got harvested according to standard protocol (scratching cells, transfer them in to | + | These two stocks got harvested according to standard protocol (scratching cells, transfer them in to 15 ml falcons, resuling 12ml solution total). After centrifugation (10 min 200g), the supernatant got transferred into two new 15 ml falcons and the pellets were resuspended with 10 ml DMEM Medium. The Stocks got freezed thawed two times so finally there were: |
<ol> | <ol> | ||
<li>DMEM as negative control</li> | <li>DMEM as negative control</li> | ||
| Line 460: | Line 460: | ||
<br /> | <br /> | ||
| - | Keep in mind, without further investigation it is not valid to say that we can purify our stocks via 10-20.000 g cantrifugation steps, because we dont know if the viral capsids are still intact! The FACS analysis are not sufficient enough to make a decision, because our last samples had YFP as GOI (=> so efficient cell sorting was not possible). We need to do qPCRs to make a valid | + | Keep in mind, without further investigation it is not valid to say that we can purify our stocks via 10-20.000 g cantrifugation steps, because we dont know if the viral capsids are still intact! The FACS analysis are not sufficient enough to make a decision, because our last samples had YFP as GOI (=> so efficient cell sorting was not possible). We need to do qPCRs to make a valid evidence. |
<br /> | <br /> | ||
<br /> | <br /> | ||
Revision as of 22:04, 2 October 2010
136. labday 01.10.2010
Test Digestions of yesterdays minipreps
Investigator: Achim, Anna
- 001_CMV: Cut with Eco, Pst & Pvu
- P654 (Lane 1)
- P655 (Lane 2)
- P686 (Lane 3)
- Control P320 (Lane 4)
- CMV_affi_VP2/3_capins-HSPG_KO: Cut with Eco & Ssp
- P680 (Lane 8)
- P681 (Lane 9)
- P684 (Lane 10)
- P685 (Lane 11)
- Control P613 (Lane 12)
- CMV_affii_VP2/3_capins: Cut with Eco & Ssp
- P678 (Lane
- P679
- Control P514
- pCerulean_Vp1up_NLS_Darpin: Cut with BamHI & XbaI
- P676 (Lane 13)
- P677 (Lane 14)
- P683 (Lane 15)
- Control P365 (Lane 20)
- Results:The expected bands should run at 500 and 4500 bp. However, just one band at ~3000 bp is visible on the gel.
Samples Stefan: Cut with Xba & Spe
- P670 (Lane 16)
- P671 (Lane 17)
- P672 (Lane 18)
- P673 (Lane 19)

mini preps of CD clones
Investigator: Kira
c(p689)= 115 ng/ul
c(690)= 151, 20 ng/ul
c(691)= 122,27 ng/ul
c(p692) = 126,42 ng/ul
test digestion of CD clones
Investigator: Kira
| Components | sample Volume/µL |
| DNA | 4,0 µl |
| BSA (10x) | 2 µl |
| Buffer no. 4 | 2,0 µl |
| Enzyme 1 XbaI | 1,0 µl |
| Enzyme 2 AgeI | 1,5 µl |
| H2O | 9,5 µl |
| Total volume | 20 |
incubation @ 37 C for approx. 2 h
1% agarose gel

Biobrick assembly pSB1C3_lITR_hTERT_beta-globin_CD
Investigator: Kira
c(pSB1C3_lITR_hTERT_beta-globin)= 333 ng/ul
c(pSB1C3_CD)= 151 ng/ul
| Components | vector Volume/µL | insert Volume/µL |
| DNA | 4,5 µl | 6 |
| BSA (10x) | 3 µl | 3 |
| Buffer no. 4 | 3,0 µl | 3 |
| Enzyme 1 XbaI | 0 µl | 1,5 |
| Enzyme 2 SpeI | 1,5 µl | 0 |
| Enzyme 3 PstI-HF | 1,0 | 1 |
| H2O | 17 | 15,5 |
| Total volume | 25 |
incubation @ 37 C for approx. 2 h
1% agarose gel

Ligation
DNA-mix: 8 ul (vector 4,6ul)+(insert 3,4 ul)
T4 ligase: 1 ul
T4 buffer: 1 ul
Incubation @ RT for 30 min
Transformation was performed according to the standard protocol w BL21 cells.
Sequencing results of pCerulean_Zegfr:1907_MiddleLinker_VP2/3_HSPG-KO
Investigator: Hanna
Comment: All N-terminal fusion approaches with VP2/3_HSPG-KO revealed positive results except of pCerulean_Zegfr:1907_MiddleLinker_VP2/3_HSPG-KO. Another clone was picked, preped, test digested and sent for sequencing.

Conclusion: Sequencing results revealed positive results.
Picking clones of pGA14_MiddleLinker_VP2/3_insCap and pGA14_MiddleLinker_VP2/3_HSPG-KO
Investigator: Hanna
Unfortunately there grew a bacteria lawn over night - it was hardly not possible to pick clones. Nevertheless I tried and picked 2 clones of pGA14_MiddleLinker_VP2/3_insCap and pGA14_MiddleLinker_VP2/3_HSPG-KO.
To do: Mini-Prep and test digestion.
Sequencing results of pSB1C3_001_VP2/3_587-KO_BAP and pSB1C3_001_VP2/3_587-KO_6xHis
Investigator: Hanna
Comment: For the creation of our super constructs, the His-Tag and the BAP motif need to be cloned into VP2/3 for N-terminal fusion to VP2. Sequencing results showed that the 6xHis and BAP motif was not cloned into VP2/3_insCap.
 |
 |
To do: Clone 587-KO_6xHis and 587-KO_BAP into pSB1C3_001_VP2/3_insCap 1. via digestion of inserts and 2. via hybridization of referring oligos - digestion of vector with 800-900 ng.
Impressions of transfection of AAV293 with pCerulean_VP1up_NLS_mVenus_VP2/3
Investigator: Adrian
Yesterday AAV293 cells were transfected with pCerulean_VP1up_NLS_mVenus_VP2/3 and GMK-TK30 as gene of interest. Today, nuclear localization of the produced mVenus_VP2/3 fusion protein was visible and can be nicely seen in the following pictures:


Conclusion: The VP2/3 fusion particle was transcribed and translated AND was transported back into the nucleus in order to be packaged by the ITR-flanked gene of interest.
Cloning of hGH_rITR into pSB1C3_lITR_phTERT_beta-globin_CFP and lITR_phTERT_beta-globin_mGMK_TK30 into pSB1C3_hGH_rITR
Investigator: Stefan
Vector name:
- pSB1C3_lITR_phTERT_beta-globin_CFP (P682)
- pSB1C3_hGH_rITR (P186)
Insert name:
- pSB1C3_hGH_rITR (P186)
- pSB1C3_lITR_phTERT_beta-globin_mGMK_TK30 (P672)
- pSB1C3_lITR_phTERT_beta-globin_mGMK_TK30 (P673)
| components | volume for P186 /µl | volume of P186 X+E /µl | volume of P672 + P673 /µl | volume of P682 /µl |
| DNA | 12 | 12 | 5 | 14 |
| BSA (10x) | 2,5 | 2 | 2 | 2 |
| Buffer 4 (10x) | 3 | 2 | 2 | 2 |
| Enzyme XbaI | 1 | 1 | - | - |
| Enzyme PstI | 1 | - | - | 1 |
| Enzyme EcoI | - | 1 | 1 | - |
| Enzyme SpeI | - | - | 1 | 1 |
| H2O | 6 | 6 | 9 | - |
| Total volume (e.g. 15,20,25,30 µl) | 25 | 25 | 20 | 20 |
Gel:
0,5 g Agarose, 50 ml TAE (1%), 3 µl GELRED , at 115 Volt

Comment: Since the first gel revealed problems with samples 672, 673 and 682 another digestion was prepared (same approach as shown above), this time using a 0,8% gel and another loading dye (without SDS).
0,4 g Agarose, 50 ml TAE (0,8%), 3 µl GELRED , at 115 Volt

Comment: Since sample 672 showed no bands this sample was discarded and cloning continued only using 673.
Gel extraction:
Was performed according to protocol.
T4 Ligation:
Ligation was performed over night.
| ligation name | 186 (X+E) + 673 | 186 + 682 |
| volume of vector | 3,51 | 4,49 |
| volume of insert | 5,38 | 2,64 |
Transformation:
Will be done tomorrow using BL21 cells.
New BL21 cells ready to use!
Investigator: Stefan
137. labday 02.10.2010
Repetition of the approach of subcloning the ViralBricks HSPG-ko BAP and His into VP2/3 with different strategies
Investigator: Bea
Comment: Unfortunately, the last experiment revealed that subcloning of the two ViralBricks did not work out which was shown by the sequencing results. Therefore we need to repeat the approach of subcloning the ViralBricks into the pSB1C3_VP2/3 which than can be used for fusing it to the n-terminal targeting approaches. Two different strategies will be carried out which means that we are digesting the pSB1C3_Viralbricks AND hybridize the oligos (BAP and His) in order to obtain some positive results.
The vector and ViralBricks were digested with BamHi and PvuII. The vector was dephosphorylated following the standard protocol.
The vector was loaded on a 1% agarose gel before dephosphorylation was conducted. The results can be seen above in the gel picture:
Result:

BioBrick Assembly of CD and mCherry
Investigator: Bea
Comment: We decided to choose some additional GOI´s for the vectorplasmid so the "new" GOI´s need to be fused to the leftITR_Promoter_betaglobin constructs. Since only one clone grew on the plate prepared by Kira where she fused the CD (cytosine deaminase) to the above mentioned construct I am going to repeat it aswell.
Digestion of the constructs:
- P689 = pSB1C3_CD
- P626 = iGEM_mCherry
- P434 = pSB1C3_leftITR_CMV_betaglobin
- P434 = pSB1C3_leftITR_phTERT_betaglobin
- P525 = pSB1C3_VP2/capins

Loading plan:
P689 (pSB1C3_CD) P626 (iGEM mCherry) P434 (pSB1C3_lITR_CMV_ß-globin) P437 (pSB1C3_lITR_phTERT_ß-globin)
Results:

After gel extraction has been performed, the ligation was carried out.
Ligation:
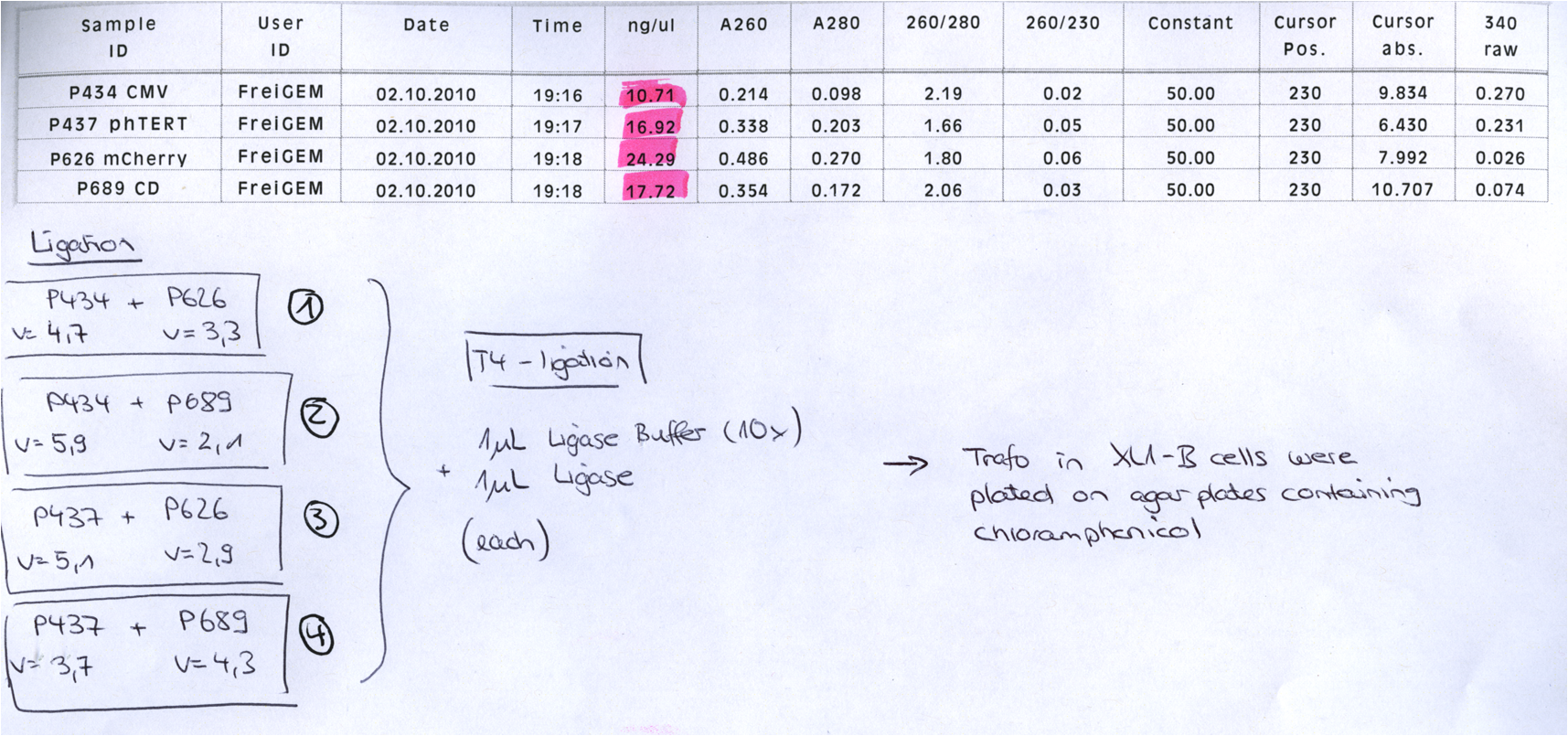
The ligation mix was transformed into XL1-Blue cells and plated on agar plates containing chloramphenicol.
Next steps:
Picking clones and perform Mini-Prep. After the Mini-Preps have been performed this construct needs to be sequenced and tested in cell culture.
comment: just one clone? uhhhh, when did you take out the plate? i put it at around 1 am..maybe they need a while to grow, no? (Kira)
Yep, I found another clone ;-) But the two look pretty good. I took them out today in the afternoon - I guess that is enough time! But if you want I can put them back into the 37° C room??! (Bea)
Mini Prep and test digestion of pGa14_MiddleLinker_VP2/3_HSPG-KO
Investigator: Hanna
Comment: In order to fuse the DARPin to the N-terminus of VP2/3_insCap and VP2/3_HSPG-KO, these motifs need to be fused to the Middle Linker (performed on 30.9.2010). In order to be able to immediately continue with cloning, BL21 cells were transformed and clones were picked in the noon. Now, the DNA needs to be preped and test digested in order to perform a 3-fragment-ligation with the DARPin and the pSB1C3_001_CMV plasmid. Unfortunately the cells of the VP2/3_insCap construct grew not densely enough. Therefore two new clones were picked from the plate and were inoculated in 10 mL LB medium. The other construct (pGa14_MiddleLinker_VP2/3_HSPG-KO) was preped and test digested.
TEST DIGESTION:
For both clones:
- DNA: 4 µL
- Buffer 4: 1 µL
- BSA: 1 µL
- EcoRI: 0.5 µL
- PstI: 0.5 µL
- H2O: 3 µL
Incubation: 1 hour.
Agarose gel: 0.8 %, 115 Volt, 1 hour.
Comment: The choice of these restriction enzymes was not clever, because if cloning didn't work, I expect one band at 2379 bp (the insert should hardly be seen) - if it worked one band at 2379 bp and a second at 2005 bp. Fortunately the bands could be separated ans revealed positive results for both clones :

Next steps:
- Mini-Prep and test digestion of pGa14_MiddleLinker_VP2/3_ins Cap.
- 3 fragment ligation of MiddleLinker_VP2/3_ins Cap or MiddleLinker_VP2/3_HSPG-KO + DARPin + pSB1C3_001_CMV backbone.
Sequencing analysis of HSPG-ko in several IRCK constructs
Investigator: Stefan
pSB1C3_001_RC_IRCK_HSPG-ko_P5tataless_RFC10 clone 1 (P687):

pSB1C3_001_RC_IRCK_HSPG-ko_P5tataless_RFC10 clone 2(P688):

pSB1C3_001_RC_IRCK_VP1-ko_HSPG-ko_P5tataless cl1 (P644):

pSB1C3_001_RC_IRCK_VP2-ko_HSPG-ko_P5tataless cl1 (P646):

Comment: Sequencing results show that all plasmids contain the HSGP-ko. Working will be continued using P644, P646 and P687.
Mini Prep of pGa14_MiddleLinker_VP2/3_inscap and pSB1C3_Darpin
Investigator: Jessica
- pSB1C3_Darpin clone1 c=158.5 ng/µl
- pSB1C3_Darpin clone2 c=205.6 ng/µl
- pGA14_MiddleLinker_VP2/3_inscap clone 1 c=147.5 ng/µl
- pGA14_MiddleLinker_VP2/3_inscap clone 2 c=121.0 ng/µl
Test-digestion:
| Components | pSB1C3_Darpin clone1/2 |
| DNA | 1,5 µl |
| BSA (10x) | 1 µl |
| Buffer no. 4 | 1 µl |
| Enzyme 1 XbaI | 0,4 µl |
| Enzyme 2 AgeI | 0,4 µl |
| H2O | 5,7 µl |
| Total volume | 10 |
Hanna wrote that a 3rd enzyme is need for the test digestion of pGA14_MiddleLinker_VP2/3_inscap but I couln't find any sequence of pGA14_MiddleLinker_VP2/3_inscap or pGA14


Test digestion of pGA14_MiddleLinker_VP2/3_inscap
Investigator: Achim
- pGA14_MiddleLinker_VP2/3_inscap clone 1 c=147.5 ng/µl (P698)
- pGA14_MiddleLinker_VP2/3_inscap clone 2 c=121.0 ng/µl (P699)
- Control: pGA14_MiddleLinker (P301)
- Digestion with EcoRI & SalI; should yield two bands of 3400 & 1000 bp. The negative control should only be cut once.

Mass Midi-Prep III
Investigator: Chris W.
Midi-Prep of:
pSB1C3_001_RC_IRCK_VP1-ko_HSPG-ko_P5tataless cl1 =P700 =B521
pSB1C3_001_RC_IRCK_VP2-ko_HSPG-ko_P5tataless cl1 =P701 =B523
pSB1C3_001_RC_IRCK_HSPG-ko_P5tataless_RFC10 clone 1 =P702 =B561
pCerulean_ZEGFR:1907_MiddleLinker_VP2/3_HSPG-KO clone 2 =P703 =B543
pCerulean_CFP_MiddleLinker_VP2/3_HSPG-KO =P704 =B507
pCerulean_ZEGFR:1907_SEG_VP2/3_HSPG-KO =P705 =B509
pCerulean_ZEGFR:1907_ShortLinker_VP2/3_HSPG-KO =P706 =B510
pCerulean_ZEGFR:1907_LongLinker_VP2/3_HSPG-KO =P707 =B511
pCerulean_6xHis_MiddleLinker_VP2/3_HSPG-KO =P708 =B512
pCerulean_VP1up_NLS_ZEGFR:1907_VP2/3_HSPG-KO =P709 =B513
pCerulean_VP1up_NLS_6xHis_VP2/3_HSPG-KO =P710 =B514
pCerulean_VP1up_NLS_mVenus_VP2/3_HSPG-KO =P711 =B515
pSB1C3_001_RC_IRCK_P5tataless clone 1 =P712 =B516
The Midi-Preps were performed according to the standard protocol yielding the following concentrations:
| plasmid-no. | P700 | P701 | P702 | P703 | P704 | P705 | P706 | P707 | P708 | P709 | P710 | P711 | P712 |
| concentration (ng/µl) | 695 | 370 | 601 | 1548 | 3996 | 3630 | 2460 | 2610 | 1860 | 2913 | 2964 | 3030 | 1200 |
Analysis of viral harvest via spectrometer
Investigator: Adrian, Patrick
The motivation: We want to figure out if it is possible to purify our viral stocks via centrifugation with 10.000 g and/or 20.000 g. In theory/ according to the literature the viral particles should be in the cells and attached to the HSPGs at the cellular surfaces.
The plan: Two different viral stocks were prepared:
Stock 1 with VP2-mVenus-fusion-Capsid loaded with TKGMK (4 wells from a 6 well plate)
- R/C 1: 50% pCerulean_VP1up_NLS_mVenus_Vp2/3 P501
- R/C 2: 50% Rep/Cap2: P449 pAAV_RC_4fachmut_VP1-ko:
- GOI: TKGMK: P82.b
- pHelper
Stock 2 "standard" virus (4 wells from a 6 well plate)
- R/C 1 P486
- GOI: TKGMK: P82.b
- pHelper
These two stocks got harvested according to standard protocol (scratching cells, transfer them in to 15 ml falcons, resuling 12ml solution total). After centrifugation (10 min 200g), the supernatant got transferred into two new 15 ml falcons and the pellets were resuspended with 10 ml DMEM Medium. The Stocks got freezed thawed two times so finally there were:
- DMEM as negative control
- resuspended Pellet with mVenus-fusioned viral capsids
- resuspended Pellet with "standard virus"
- supernatant with mVenus-fusioned viral capsids
- supernatant with "standard virus"
The fluorescence of each stock was measured via spectrometer. After that, stocks 2 and 4 were centrifugated 10min with 10.000 g followed by an other fluorescence measuring. Finally the stocks got spin down with 20.000 g for 10 min
The results:
The conclusions: The highest amount of fluorescence was measured in the supernatant, the individual centrifugation steps had no decrease in fluorescence as consequence!
Keep in mind, without further investigation it is not valid to say that we can purify our stocks via 10-20.000 g cantrifugation steps, because we dont know if the viral capsids are still intact! The FACS analysis are not sufficient enough to make a decision, because our last samples had YFP as GOI (=> so efficient cell sorting was not possible). We need to do qPCRs to make a valid evidence.