<partinfo>BBa_K300000</partinfo> - BioBrick integrative base vector for E. coli
Materials and Methods
Plasmids and strains: the <partinfo>BBa_J72008</partinfo> helper plasmid was kindly given by Prof. JC Anderson (UC Berkeley). BW23474 (<partinfo>BBa_K300985</partinfo>), MC1061 (<partinfo>BBa_K300078</partinfo>) and MG1655 (<partinfo>BBa_V1000</partinfo>) E. coli strains and the pCP20 helper plasmid were purchased from the Coli Genetic Stock Center (Yale University).
Verification primers: all the oligonucleotides were purchased from Primm (San Raffaele Biomedical Science Park, Milan, Italy). The P1 (<partinfo>BBa_K300975</partinfo>) and P4 (<partinfo>BBa_K300978</partinfo>) primers had already been used in [Anderson JC et al., 2010]. The P2 (<partinfo>BBa_K300976</partinfo>) and P3 (<partinfo>BBa_K300977</partinfo>) primers have been newly designed using ApE and Amplify 3X. P2 and P3 have been designed also considering the previously used verification primers P2 and P3 in the pG80ko integrative plasmid, described in [DeLoache W, 2009].
The relative position of the P1, P2, P3 and P4 primers is shown in Fig.1:
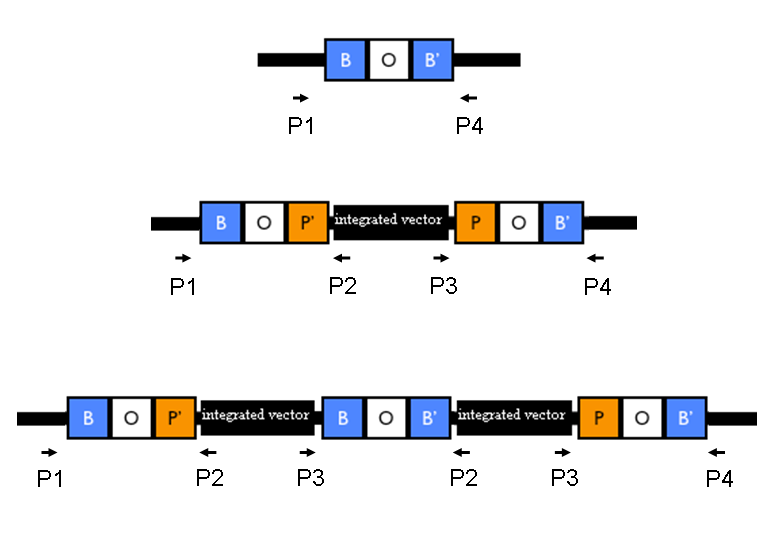 Figure 1: Relative position of the verification primers. a) no integrants; b) single integrant and c) integrant with multiple tandem copies. P1/P2 and P3/P4 pairs give an amplicon when at least one copy of the vector is integrated in the Phi80 locus. P2/P3 pair show an amplicon only when multiple tandem copies occur. |
Competent cells preparation: all the E. coli strains were made competent following a slightly modified version of the protocol described in [Sambrook J et al., 1989]. Briefly, cells were grown to and OD600 of ~0.4-0.6, harvested (4000 rpm, 10 min, 4°C) and the supernatant discarded. Cells were resuspended in (30 ml for each 50 ml of initial culture) pre-chilled Mg-Ca buffer (80 mM MgCl2, 20 mM CaCl2), centrifuged as before and the supernatant discarded. Cells were resuspended in (2 ml for each 50 ml of initial culture) pre-chilled Ca buffer (100 mM CaCl2, 15% glycerol), aliquoted in 0.5 ml tubes and freezed immediately at -80°C. Test the transformation efficiency in Colony Forming Units (CFU)/ug of transformed DNA
The Chloramphenicol concentration in plates was 34 ug/ml for the high copy plasmids, 12.5 ug/ml for the medium/low copy plasmids and 12.5 for the three control strains transformed with the R6K plasmid.
Integration protocol:
- Transform the <partinfo>BBa_J72008</partinfo> helper plasmid in the host strain (MC1061 or MG1655) and select transformants on Amp (50 ug/ml) plates under permissive conditions (30°C) overnight.
- Inoculate a single colony in selective LB and let the culture grow at 30°C, 220 rpm. When the culture reaches the OD600 of 0.4-0.6 prepare chemically competent cells.
- Transform the integrative vector with the desired insert in the BBa_J72008-containing strain and select co-transformants on Cm (34 ug/ml) plates under permissive conditions (30°C) overnight. At this temperature <partinfo>BBa_J72008</partinfo> can be replicated and so the pir protein product can be expressed in the cells. The pir product enables the propagation of the integrative vector by replicating the R6K origin.
- Inoculate a single colony in 5 ml of LB + Cm at 12.5 ug/ml and incubate the culture at 37°C, 220 rpm overnight. At this temperature the <partinfo>BBa_J72008</partinfo> helper cannot be replicated and the Phi80 integrase is expressed by the remaining copies of the helper. The bacteria that are able to grow in this selective medium should be correct integrants because the integrative vector cannot be replicated by the pir product anymore.
- Streak the culture on a Cm plate (at 12.5 ug/ml) and incubate it at 43°C overnight to ensure the loss of the helper plasmid. The bacteria that form colonies should be correct integrants without the <partinfo>BBa_J72008</partinfo> helper plasmid.
Validate the loss of the helper plasmid by inoculating colonies in Cm (at 12.5 ug/ml) media and counterselecting them in Amp (at 50 ug/ml) media. Validate the correct integration position by performing colony PCR with primers P1/P2, P3/P4, P1/P4, P2,P3 and VF2/VR. Validate the phenotype (when possible).
Expected amplicon length [bp] when the vector is integrated into the Phi80 locus:
|
| No integrant
| Single integrant
| Multiple tandem integrants (>1)
|
| VF2/VR
| none
| 280 + insert length
| 280 + insert length
|
| P1/P4
| 546
| 546 + insert length + 2171 (i.e. the BBa_K300000 length)
| 546 + insert length + 2171 (i.e. the BBa_K300000 length)
|
| P1/P2
| none
| 452
| 452
|
| P3/P4
| none
| 666
| 666
|
| P2/P3
| none
| none
| 572
|
Marker excision protocol:
- Inoculate an integrant in selective LB medium and let it grow to OD600=0.4-0.6. Prepare chemically competent cells.
- Transform the pCP20 helper plasmid in the competent strain and select transformants on Amp (100 ug/ml) plates under permissive conditions (30°C) overnight. At this temperature the pCP20 can be replicated. The pCP20 plasmid contains Amp and Cm resistance markers, a thermoinducible Flp recombinase expression system and a heat-sensitive replication origin. The permissive temperatures for the pCP20 propagation are the same as <partinfo>BBa_J72008</partinfo>.
- Inoculate a single colony in 5 ml of LB without antibiotic and incubate the culture at 37°C, 220 rpm overnight. At this temperature the pCP20 helper cannot be replicated and the Flp recombinase is expressed by the remaining copies of the helper. The bacteria should loose the R6K origin and the Cm resistance upon FRT sites recombination, mediated by Flp.
- Streak the culture on a LB plate and incubate it at 43°C overnight to ensure the loss of the helper plasmid. The bacteria that form colonies should be without the pCP20 helper plasmid.
Validate the loss of the helper plasmid by inoculating colonies in Amp (at 100 ug/ml) media and validate the loss of the Cm resistance from the genome by inoculating colonies in Cm (at 12.5 ug/ml) media. Validate the correct length of the integrated part without Cm resistance and R6K origin by performing colony PCR with primers P1/P4 (which amplify the entire Phi80 locus) and VF2/VR (which amplify the integrated part). Validate the phenotype (when possible).
Colony PCR: a single colony or 1 ul of culture was added to the Invitrogen Platinum Taq reaction mix and was heated at 94°C for 10 min. Then it was assayed with this cycle (X 35): 94°C 30 sec, 60°C (for VF2/VR) or 63°C (for the other primers) 30 sec, 72°C according to the amplicon expected length (1Kb/min). Then the reaction was kept at 72°C for 10 min and it was run on a 1% agarose gel with the GeneRuler 1Kb Plus DNA ladder (Fermentas).
Fluorescence assays: integrants were inoculated in 1 ml of M9 + Cm (12.5 ug/ml) and grown at 37°C, 220 rpm overnight. The cultures were diluted 1:100 in 2 ml of selective M9 and let grow for about 4-6 hours under the same conditions as before. Three 200 ul aliquots for each culture were transferred to a 96-well microplate and assayed in the Infinite F200 microplate reader (Tecan) for about 20 hours with the following kinetic cycle: 37°C, 5 min sampling time, linear shaking 15 sec (amplitude=3), wait 5 sec, measure absorbance at 600nm, measure fluorescence with the proper filter (EX:nm/EM:540nm for GFP or EX:535nm/EM:620nm for RFP) with gain=70. The same protocol was followed for the MC1061 and the MG1655 non-integrant strains, which were grown in M9 without antibiotic.
Data analysis: the absorbance measurements were normalized by subtracting the absorbance of the M9, while the fluorescence measurements were normalized by subtracting the fluorescence of the non-integrant strains over time. For each well, the Scell signal (proportional to the reporter protein synthesis rate per cell) was computed as (1/OD600*dXFP/dt), where OD600 is the normalized absorbance and XFP is the normalized fluorescence. The Scell signal was then averaged over time to obtain a single value for each well. Results are presented as the average Scell with their 95% confidence intervals of the mean.
Results
Integration of the desired BioBrick part into the Phi80 genome locus
MC1061 and MG1655 were chosen as host strains for integration. <partinfo>BBa_K173001</partinfo> (constitutive strong promoter with GFPmut3) and the EcoRI-PstI fragment of <partinfo>BBa_J61002</partinfo>-<partinfo>BBa_J23101</partinfo> (here called PconRFP - constitutive strong promoter with RFP) were chosen as two proof of concept BioBrick parts to test the integration capability of the <partinfo>BBa_K300000</partinfo> vector in the Phi80 genome locus of these strains. For this reason, <partinfo>BBa_K173001</partinfo> and PconRFP were ligated in <partinfo>BBa_K300000</partinfo> (digested with EcoRI-PstI) and propagated using BW23474.
The integration protocol was performed as described in the Materials and Methods section for 4 different combination:
| Integrant name
| Strain
| Insert of <partinfo>BBa_K300000</partinfo>
|
| MC-GFP
| MC1061
| <partinfo>BBa_K173001</partinfo>
|
| MC-RFP
| MC1061
| PconRFP
|
| MG-GFP
| MG1655
| <partinfo>BBa_K173001</partinfo>
|
| MG-RFP
| MG1655
| PconRFP
|
Three colonies grown after the overnight incubation at 43°C (step 5 of integration protocol) were analyzed for each plate. These 12 clones were called: MC-GFP-A,B,C , MC-RFP-A,B,C , MG-GFP-A,B,C and MG-RFP-A,B,C.
Validation of the loss of BBa_J72008: all the picked colonies did not grow in Amp (50 ug/ml) media, thus validating that <partinfo>BBa_J72008</partinfo> Amp-resistant helper had been actually cured from the cells. However, one of these 12 clones (MG-GFP-A) also failed to grow in Cm (12.5 ug/ml) liquid media, probably because of a mistake in its inoculation. We decided not to consider this clone and to continue with 11 clones.
Validation of the actual integration site: colony PCR was performed for all the 11 clones, using MC1061 and MG1655 without integrants as negative controls. Primer pairs P1/P2 and P3/P4 were used to validate the presence of the integrative vector in the Phi80 genomic locus, while the primer pair P2/P3 was used to validate the presence of multiple tandem integrants (see Fig.1 in Materials and Methods).
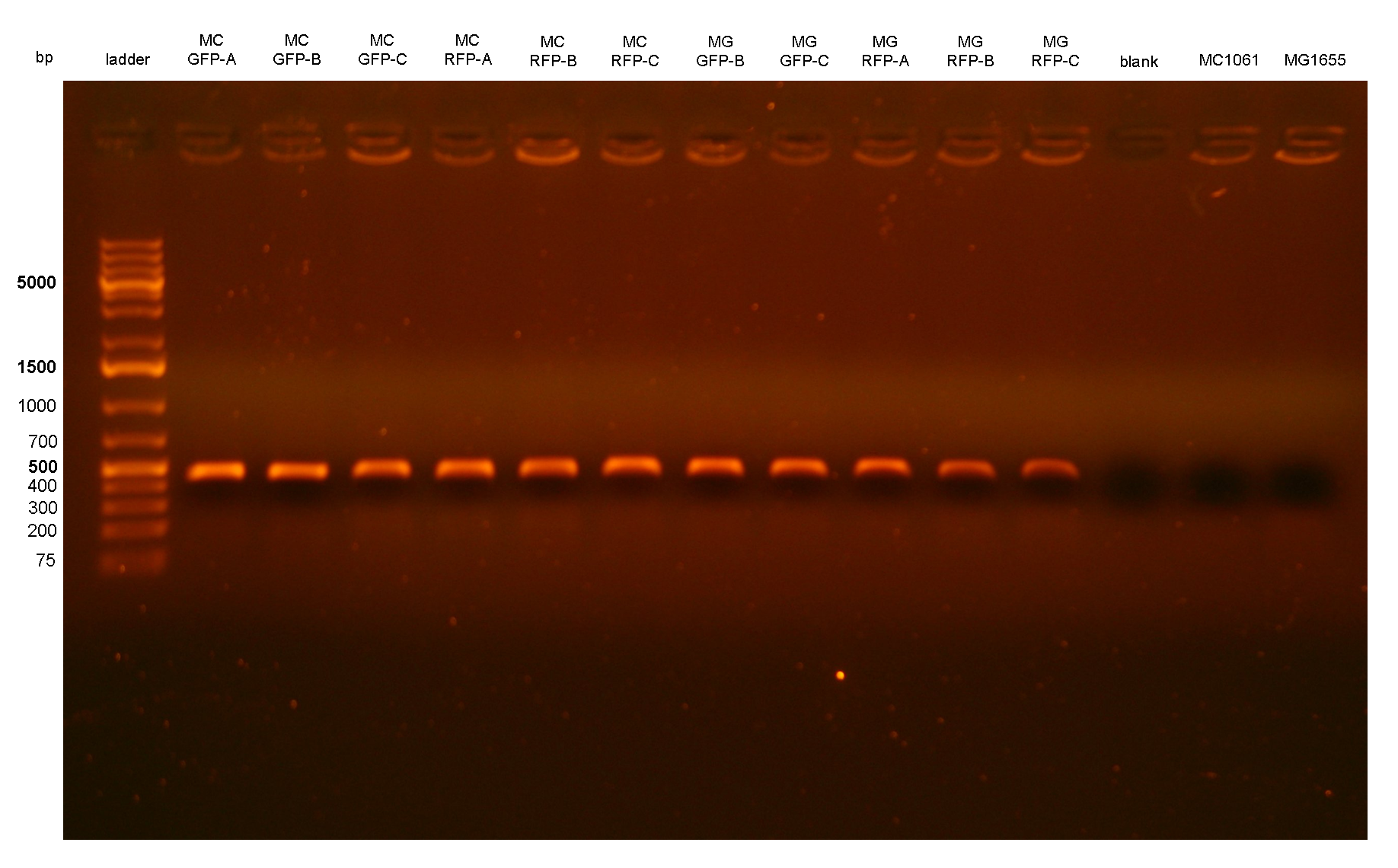 Figure 2: colony PCR with P1/P2 on all the 11 integrant clones. The blank is the reaction mix without bacteria. Expected amplicon for correct integrants: 452 bp. |
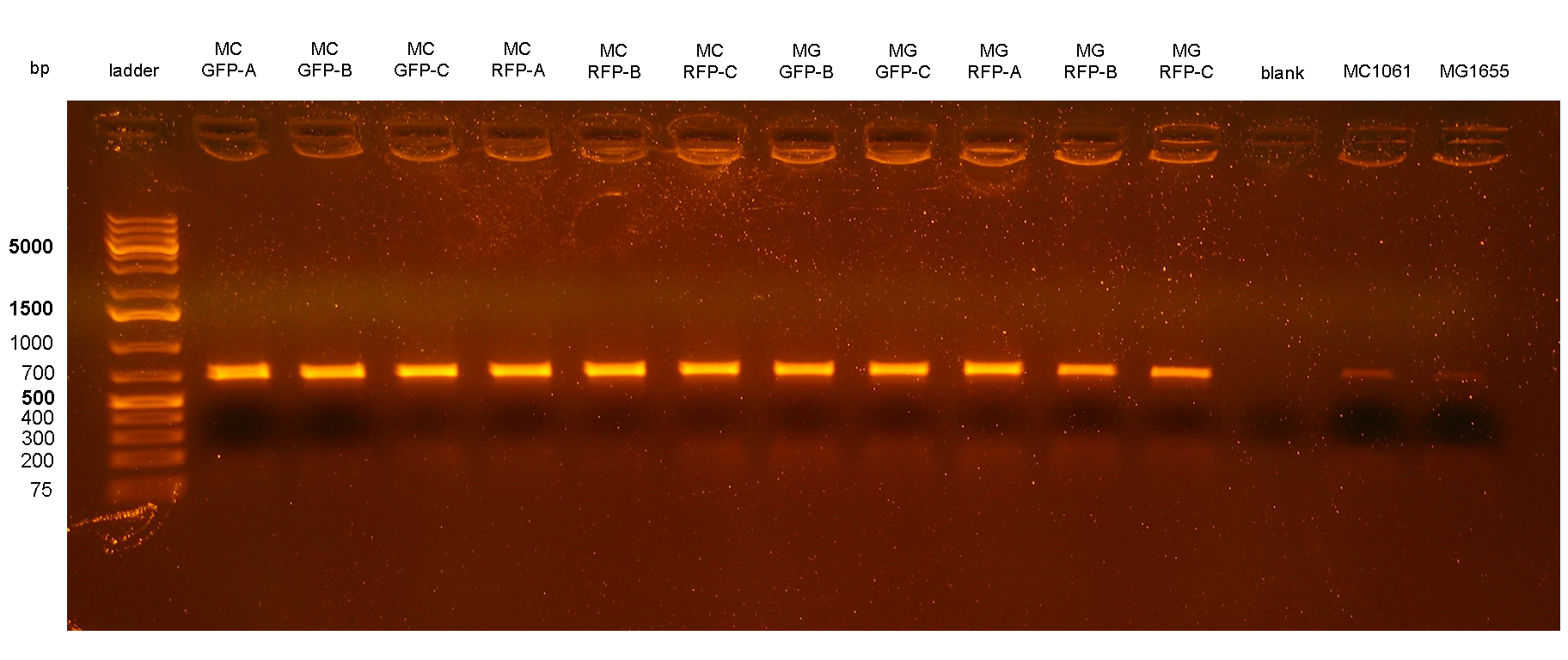 Figure 3: colony PCR with P3/P4 on all the 11 integrant clones. The blank is the reaction mix without bacteria. Expected amplicon for correct integrants: 666 bp. |
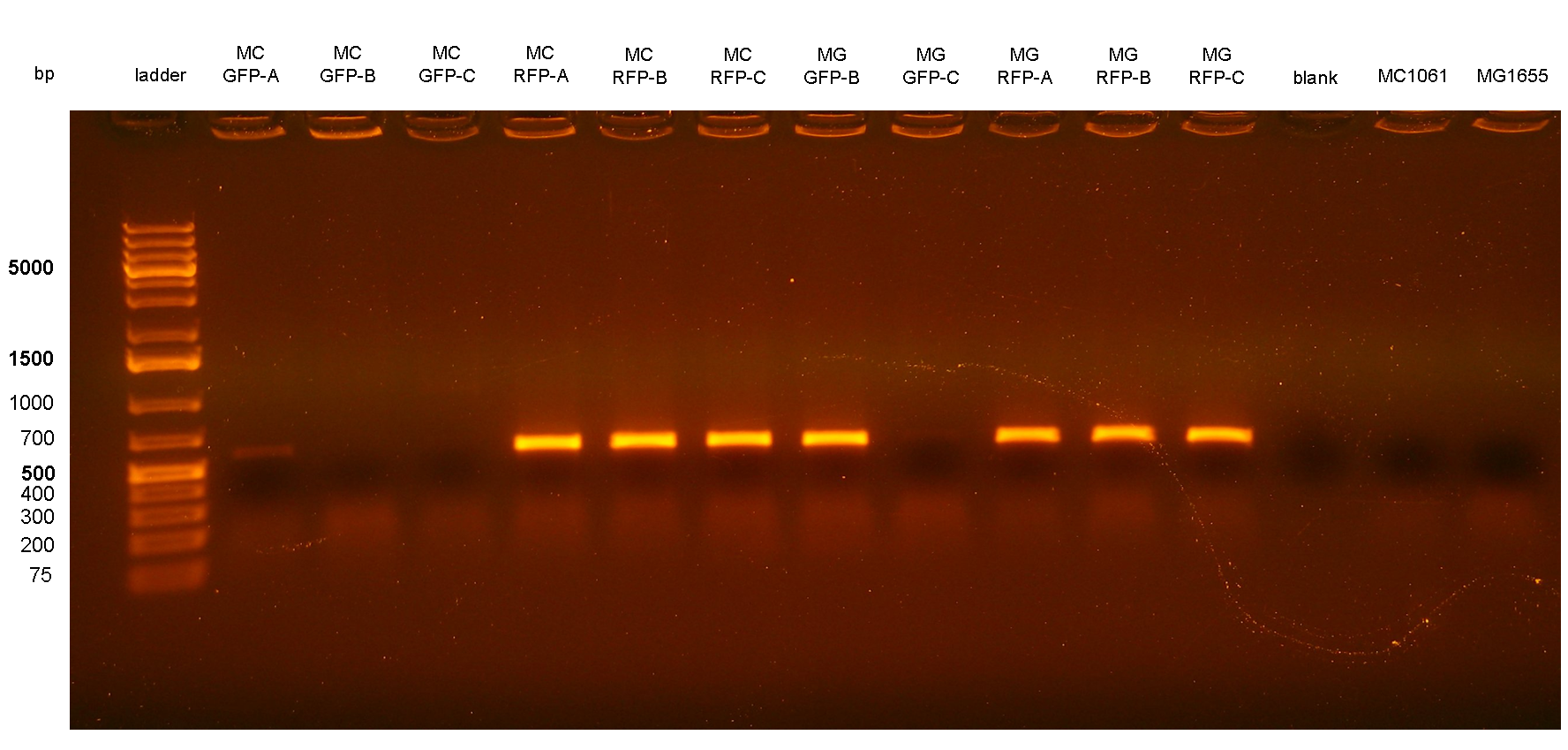 Figure 4: colony PCR with P2/P3 on all the 11 integrant clones. The blank is the reaction mix without bacteria. The lanes with the amplicon were expected to come from bacteria with multiple tandem integrants. Expected amplicon for multiple integrants: 572 bp. |
PCR results with primers P1/P2 and P3/P4 showed that each clone had the correct integrant in the correct genomic position (see Materials and Methods for a list of the expected amplicon lengths). Negative controls showed no amplicons with primers P1/P2 as expected, but showed an unexpected band with P3/P4. The reason of the presence of this band was not further investigated and the results with this primer pair cannot be a useful tool for future analysis. Anyway, the P1/P2 primer pair can be sufficient to successfully validate the presence of the DNA of interest in the Phi80 genomic locus.
PCR results with primers P2/P3 showed that two clones (MC-GFP-B and MC-GFP-C) were single integrants, while all the other clones were multiple tandem integrants (i.e. the Phi80 locus contained more than one copy of the DNA of interest). Negative controls showed no amplicons, as expected.
Validation of the integrants phenotype: all the 11 clones were assayed as described in the Materials and Methods section. Unfortunately, the green fluorescent clones (MC-GFP-A,B,C and MG-GFP-B,C) did not show appreciable differences when compared to negative controls, most probably because the autofluorescence of the cells was too high and hid the GFP signal. For this reason, GFP clones were not considered for further analysis. Other instruments should be used to detect the GFP signal.
On the other hand, RFP clones (MC-RFP-A,B,C and MG-RFP-A,B,C) all showed a higher fluorescence than the negative controls (see Fig.5). As Fig.5 show, the fluorescence of the three MG-RFP had a higher variability between clones when compared to the three MC-RFP. However, the clones were not necessarily expected to behave in the same way because all of them were multiple tandem integrants and the copy number of the PconRFP construct could be arbitrary.
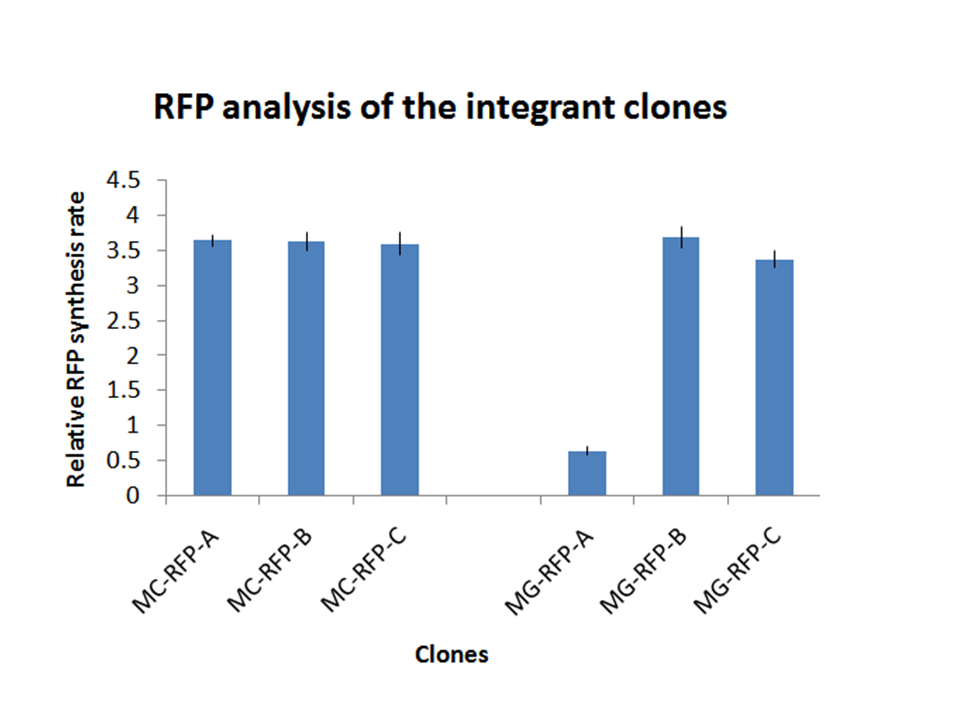 Figure 5: relative RFP synthesis rate for all the RFP expressing clones. Note: as a reference, typical values of the relative RFP synthesis rate measured for PconRFP in a low copy vector (~5 plasmids per cell) are about 6-7 fold higher (data not shown). |
Chloramphenicol resistance marker excision
The marker excision was performed on two of the previously validated integrant strains: MC-RFP-A and MG-RFP-A (even if they were multiple tandem integrants).
The marker excision protocol was performed as described in the Materials and Methods section for both strains, here named:
| Original name
| Name after marker excision
|
| MC-RFP
| MC-RFPflip
|
| MG-RFP
| MG-RFPflip
|
Three colonies grown after the overnight incubation at 43°C (step 4 of marker excision protocol) were analyzed for each plate. These 6 clones were called MC-RFPflip-A,B,C and MG-FRPflip-A,B,C.
Validation of the loss of pCP20 and the resistance marker: all the 6 picked colonies failed to grow on both Amp (100 ug/ml) media and Cm (12.5 ug/ml) media. They could only grow in LB without antibiotics, thus validating that the pCP20 helper had been actually cured and the R6K-CmR DNA containing the Chloramphenicol selection marker had been actually eliminated.
Validation of the length of the integrated part: colony PCR was performed for all the 6 clones, using MC1061 and MG1655 without integrants as negative controls. Primer pairs VF2/VR and P1/P4 were used to validate if the passenger of interest was still present in the genome and the length of the entire Phi80 locus respectively after the marker excision.
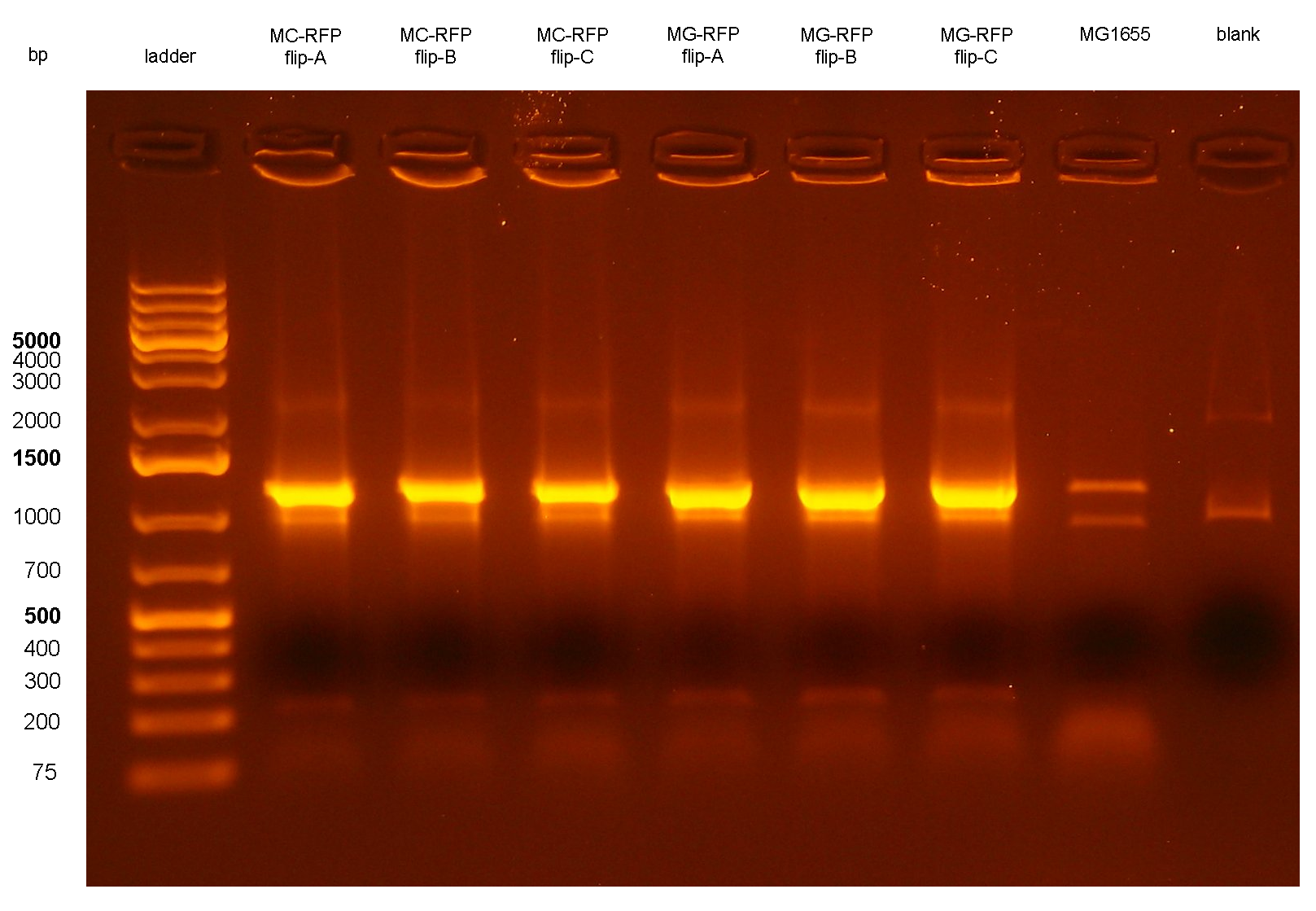 Figure 6: colony PCR with VF2/VR on all the 6 flipped clones. The blank is the reaction mix without bacteria. Expected amplicon for correct insert: 1.2 Kb. |
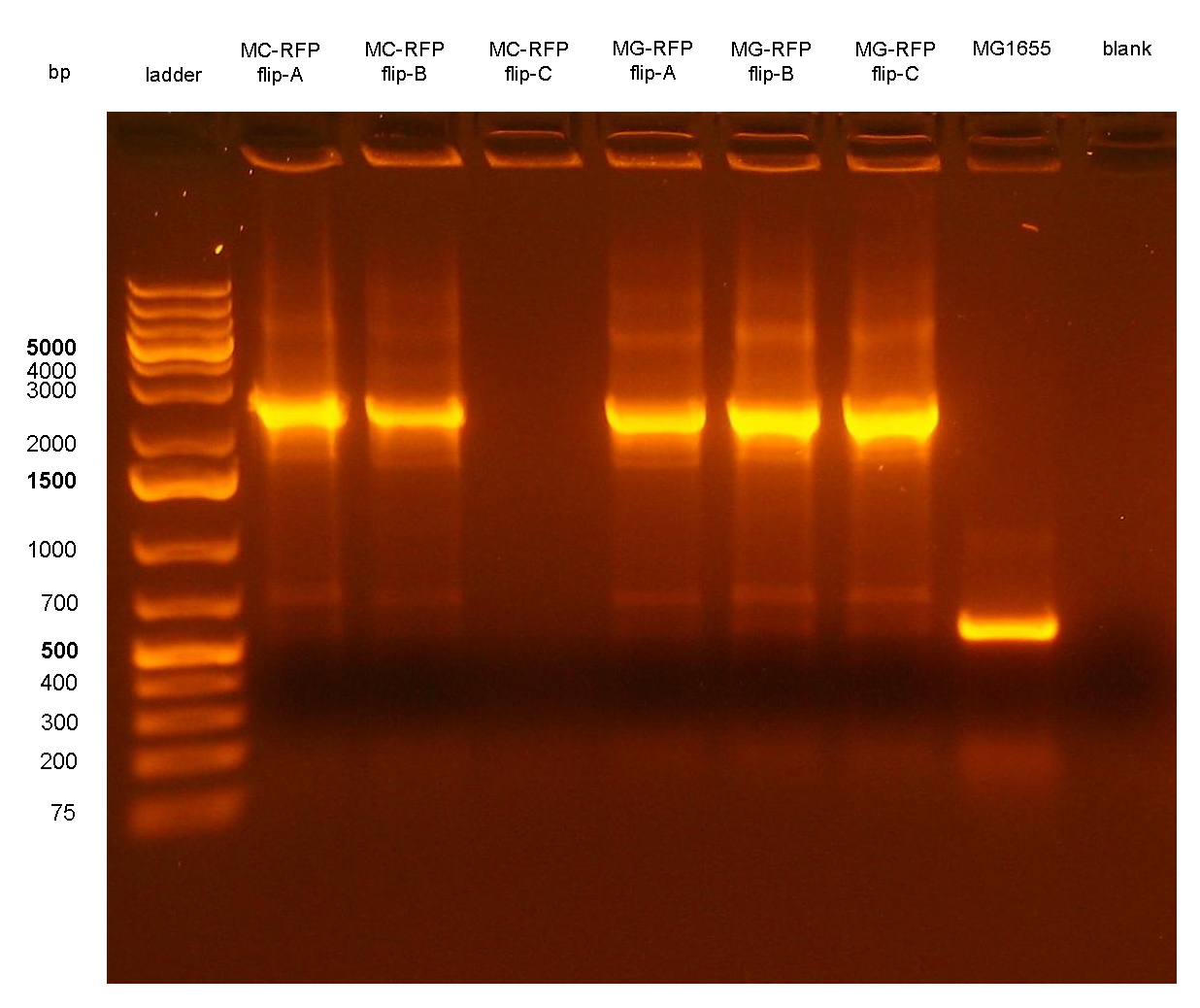 Figure 7: colony PCR with P1/P4 on all the 6 flipped clones. The blank is the reaction mix without bacteria. Expected amplicon for correct construct in the correct position: 2.3 Kb. Expected amplicon for the non-integrant strain MG1655: 546 bp. |
PCR results with primers VF2/VR showed that all the 6 clones still contain the passenger of interest, i.e. PconRFP, in the genome after the marker excision. The reaction blank, the MG1655 strain (neg control) and also the other samples showed some extra bands, but the ~1.2Kb amplicons of MC-RFP-A,B,C and MG-RFP-A,B,C had the correct length and was much brighter than the other bands.
PCR results with primers P1/P4 (Fig.7) showed that an amplicon of ~2.3Kb was present in all but one screened clones, while the MG1655 negative control showed the expected 546bp length for a non-integrant. MC-RFPflip-C did not show the P1-P4 amplicon because the reaction failed: the tube was damaged and the reaction mix was completely evaporated at the end of the PCR program. For this reason, a PCR was performed again on this clone (Fig.8).
The ~2.3Kb amplicon was consistent with a single integrant of <partinfo>BBa_K300000</partinfo>-PconRFP without the R6K-CmR DNA fragment, thus validating the successful excision of the FRT-flanked DNA fragment containing R6K-CmR and confirming that PconRFP was still present in the correct locus in single copy.
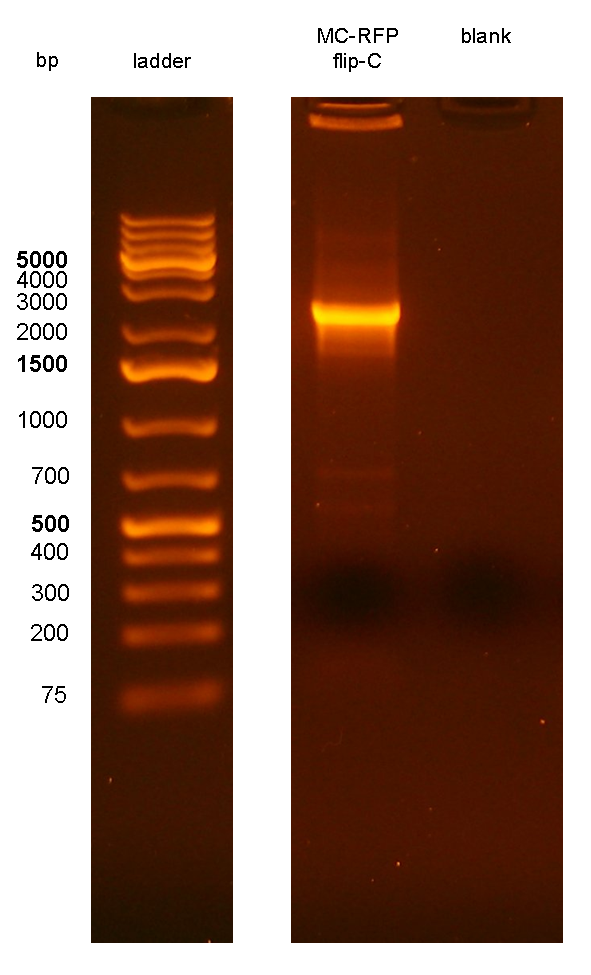 Figure 8: colony PCR with P1/P4 on MC-RFPflip-C clone. The blank is the reaction mix without bacteria. Expected amplicon for correct construct in the correct position: 2.3 Kb. |
These results showed that, even if the clones were multiple tandem integrants, they became single integrants after marker excision. This is because the Flp recombinase mediated the recombination of all the FRT sites of the multiple integrants until only a single FRT site was present in the Phi80 locus, thus leaving only the single integrant of interest without the selection marker in the genome.
Validation of the marker-less phenotype:all the 6 clones were assayed as described in the Materials and Methods section. They all showed a low variability and their fluorescence was lower than their two parents, i.e. MC-RFP-A for the MC1061 strains and MG-RFP-A for the MG1655 strains (see Fig.9). This result is consistent with the copy number of the PconRFP construct in the clones, in fact both MC-RFP-A and MG-RFP-A were multiple tandem integrants, while MC-RFPflip-A,B,C and MG-RFPflip-A,B,C were single integrants, as described above.
All the MG-RFPflip showed a very low relative RFP synthesis rate when compared to the other strains, but the signal is systematically grater than the fluorescence of the negative control, thus validating the phenotype for the MG1655 strain. MC-RFPflip-A,B,C showed a higher fluorescence than MG-RFPflip-A,B,C.
In conclusion, it has been demonstrated that, even after the marker excision process, the phenotype of the engineered cells is maintained.
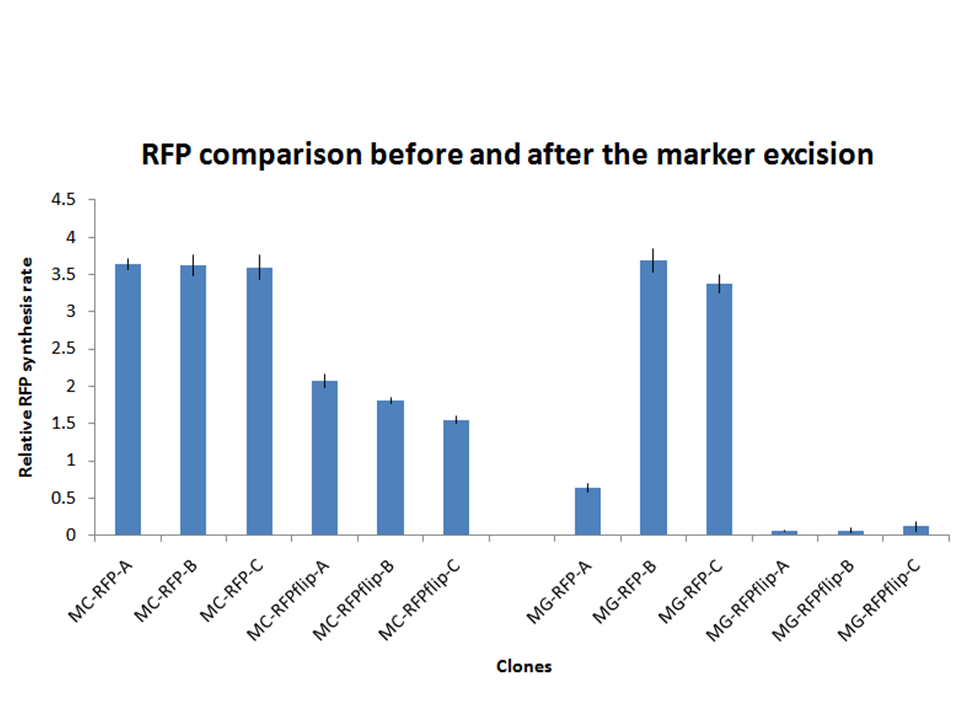 Figure 9: relative RFP synthesis rate for all the RFP-expressing clones after marker excision. In this figure, the bars corresponding to the fluorescence of the clones before marker excision is also reported to facilitate the comparison between them. Note that all the three flip clones are derived from MC-RFP-A for the MC1061 clones and from MG-RFP-A for the MG1655 clones. |
Discussion
A novel integrative vector for E. coli has been successfully designed, constructed and used to integrate two proof of concept protein expression systems in two commonly used E. coli strains.
The results showed that the vector is fully functional and can integrate into the correct targeted locus of the host chromosome through the Phi80 site-specific recombination system by using <partinfo>BBa_J72008</partinfo>, an existing BioBrick helper plasmid from the Registry. In most cases, the integration occurs in tandem copies, probably because of the too high Chloramphenicol concentration used during the selection of integrants, which forces multiple integration of Cm-resistant constructs. This concentration was the same used during the pSC101 low copy plasmid (~5 copies per cell) selection. In some cases, it is desirable to have a single copy of the desired BioBrick in the genome, for example when the gene dosage is important. In [Haldimann A and Wanner BL, 2001] the usage of Chloramphenicol at 6 ug/ml yielded a very high percentage of single integrants. However, when tested in our lab, the MG1655 strain could survive on LB plates with Cm at 6 ug/ml and also at 8 ug/ml. For this reason a higher concentration of Cm was chosen for selection. Further studies should investigate the optimal antibiotic concentration to yield the highest single integrants percentage as possible.
The Flp/FRT mediated excision of the R6K and, most importantly, of the Cm resistance marker also worked by using the pCP20 helper plasmid. The estimated efficiency of this process was 100%. In addition, multiple tandem integrants became single integrants after the marker excision. This is because the Flp recombinase mediated the recombination of all the FRT sites of the multiple integrants until only a single FRT site was present in the Phi80 locus. The marker excision is a powerful tool to engineer microbial strains for industrial protein manufacturing because the engineered organism should not carry unsafe antibiotic resistances that may be diffused in the environment.
The fluorescence phenotype confirmed the correct integration into the E. coli chromosome. As expected, in general multiple integrants showed a higher fluorescence than the single integrants.
The BioBrick compatibility and the vector modularity give the possibility to the scientific community to stably engineer novel biological functions in E. coli with a very easy and user friendly methodology. A user’s handbook about the vector usage is shared in the Registry, as well as the users experiences and the compatibility information.
<partinfo>BBa_K300001</partinfo> - BioBrick integrative base vector for S. cerevisiae
The integration capability of this vector has been tested in S288C S. cerevisiae strain (<partinfo>BBa_K300979</partinfo>). Here is reported the followed protocol and the obtained results.
Protocol:
- S288C strain (Open Biosystems) was inoculated in 5 ml of YPD from a long term 15% glycerol stock and grown for 24h (30°C, 200rpm).
- The culture was diluted 1:10 in 50 ml of pre-warmed YPD in a 250 ml flask and was grown for additional 4 hours under the same conditions as before.
- Cells were pelleted (4000 rpm, 5 min) and resuspended in 25 ml of deionized water.
- Cells were pelleted (4000 rpm, 5 min), the supernatant was discarded and the pellet was resuspended in 1 ml of deionized water and transferred into a 1.5 ml tube.
- Cells were pelleted (4000 rpm, 30 sec), the supernatant was discarded and the pellet was resuspended in deionized water to a final volume of 1 ml (vortex mix vigorously).
- Three 100 ul aliquots were transferred into 1.5 ml tubes, while the remaining 600 ul of cells were not used in this protocol.
- The three tubes were centrifuged (4000 rpm, 30 sec) and the supernatant discarded.
- Each of the three pellets were resuspended (vortex mix vigorously) in 360 ul of transformation mix (240 ul of PEG 3350 50% w/v, 36 ul of LiAc 1.0 M, boiled salmon sperm DNA, 34 ul of linearized plasmid DNA plus water). The salmon sperm DNA was boiled for 5 min and pre-chilled before adding it in the transformation mix. The plasmid DNA was previously digested with SbfI (Fermentas), purified with the NucleoSpin Extract II kit (MN) and quantified with the NanoDrop in order to add 1 ug of DNA to the transformation mix.
- The tubes were heated at 42°C for 40 min.
- Cells were pelleted (4000 rpm, 30 sec), the supernatant was removed by pipetting and the pellet was gently resuspended in 1 ml of deionized water.
- Cells were pelleted (4000 rpm, 30 sec), the supernatant was discarded, the pellet was resuspended in 1 ml of YPD and incubated at 30°C, 200 rpm for 3 hours.
- Cells were pelleted (4000 rpm, 30 sec), resuspended in 200 ul of YPD and plated on a YPD agar plate with G418 antibiotic at 200 ug/ml.
- The plates were incubated at 30°C for about 3 days until colonies appeared.
The integration efficiency was estimated as the colony forming units (CFUs) yielded for each ug of DNA.
Protocol references:
[1] http://openwetware.org/wiki/High_Efficiency_Transformation
[2] Guldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH (1996), A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Research, Vol. 24, No. 13 2519–2524.
Results:
The transformed inserts and their integration efficiency in S288C are listed here:
| SbfI-digested plasmid
| ug of transformed DNA
| # of colonies
| Estimated integration efficiency [CFU/ug]
|
| <partinfo>BBa_K300001</partinfo>-<partinfo>BBa_K300006</partinfo>
| 1
| 1700
| 1.7*10^3
|
| <partinfo>BBa_K300001</partinfo>-<partinfo>BBa_K300007</partinfo>
| 1
| 6500
| 6.5*10^3
|
| no DNA
| 0
| 0
| 0
|
These results suggest that the integrative vector actually works and that the selection marker is highly specific (no colonies appeared on the "no DNA" plate).
The correct phenotype of the S288C bearing these parts has still to be validated (by mOrange fluorescence measurement for the <partinfo>BBa_K300007</partinfo> part), as well as the actual integration position (by PCR).
<partinfo>BBa_K300004</partinfo> - Engineered pH-inducible intein (codon optimized for E. coli) - internal domain
<partinfo>BBa_K300010</partinfo> - PoPS-based self-inducible device
The PoPS-based self-inducible device should be assembled downstream of a promoter and upstream of the gene of interest to obtain a device able to start the production of the target protein at a pre-determined user-defined culture density.
LuxI encodes a protein responsible for the synthesis of 3-oxo-C6-homoserine-lactone (3OC6HSL or simply HSL) and its production should be regulated by a different promoter, depending on the deisred O.D.start.
luxR encodes a protein capable to bind the HSL and is constitutively expressed under the control of pTetR promoter (<partinfo>BBa_R0040</partinfo>).
The features of this device have been evaluated in many different experimental conditions:
- varying the strength of the promoter controlling the production of the signal molecule (Sender Modulation)
- varying the copy number of vectors bearing both Sender and Receiver circuits
- varying the growth medium (LB or M9)
The results obtained are reported in the sections below.
<partinfo>BBa_K300093</partinfo>, <partinfo>BBa_K300094</partinfo>, <partinfo>BBa_K300097</partinfo>, <partinfo>BBa_K300095</partinfo> and <partinfo>BBa_K300084</partinfo> - Phasin and Intein-based tags for protein purification
These parts are built by assembling Phasins (<partinfo>BBa_K300002</partinfo> and <partinfo>BBa_K300003</partinfo>) that are able to bind to PolyHydroxyAlkanoates (PHA)
granules and Intein (<partinfo>BBa_K300004</partinfo>), a sequence capable of self-exciding from a precursor protein through a process known as self-splicing. In addition a flexible linker sequence (<partinfo>BBa_K105012</partinfo>) has been used to connect these parts in order to facilitate the binding and folding of the tag and the target protein of interest. Thanks to Phasin and Intein properties these parts can be used as high-specific TAGs for low-cost protein purification.
At the moment were were not able to test Intein efficiency, but we could check if they affected the right folding of the target protein: we achieved this goal through the Silver Standard Assembly by using the GFP (<partinfo>BBa_K300005</partinfo>).
These parts has been characterized respectively through:
- pTet costitutive promoter devices:
- <partinfo>BBa_K300088</partinfo>
- <partinfo>BBa_K300090</partinfo>
- <partinfo>BBa_K300099</partinfo>
- 3OC6HSL inducible devices:
- <partinfo>BBa_K300091</partinfo>
- <partinfo>BBa_K300092</partinfo>
and compared to a positive (<partinfo>BBa_K173000</partinfo>) and negative (<partinfo>BBa_B0031</partinfo>) control.
pTet costitutive promoter devices
Methods
Inoculum (into 5 ml LB+Amp) from glycerol stock of:
- <partinfo>BBa_K300088</partinfo>
- <partinfo>BBa_K300090</partinfo>
- <partinfo>BBa_K300099</partinfo>
- <partinfo>BBa_K173000</partinfo> (positive control)
- <partinfo>BBa_B0031</partinfo> (negative control)
They were let grow ON at +37°C, 220 rpm.
The following day cultures were diluted 1:100 and let grow again for about five hours at +37°C, 220 rpm.
Optical density (O.D.) of each cell culture was than measured with TECAN Infinte F200. Samples were diluted in order to obtain the same O.D. equal to 0,02.
Than we performed a 21 hours' experiment with measurements of absorbance and green fluorescence every five minutes with TECAN Infinite F200. Each value shown is the mean of three measurements, from GFP data that of a non-fluorescent culture (negative control) was subtracted; cultures were shaken for 15 seconds every five minutes.
Results
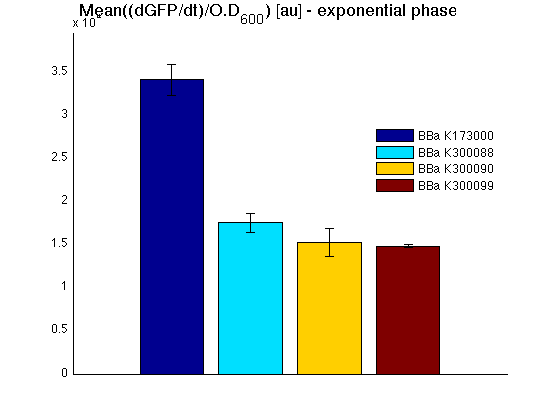 Mean (dGFP/dt)/O.D. over the exponential phase (under the hypothesis that GFP half-life in fusion contructs is similar to the original one - <partinfo>BBa_E0040</partinfo>) |
| Culture | Doubling time [min.] |
| <partinfo>BBa_K173000</partinfo> | 77 |
| <partinfo>BBa_K300088</partinfo> | 75 |
| <partinfo>BBa_K300090</partinfo> | 76 |
| <partinfo>BBa_K300099</partinfo> | 76 |
| <partinfo>BBa_B0031</partinfo> | 69 |
Discussion
All cell cultures showed a similar growth curve and doubling time was computed as described here in order to have informations about the burden due to synthesis of such fusion proteins. It's possible to see that all doubling time are very similar; it's possible to assert that the expression of these BioBrick parts doesn't cause abnormal stress to the cells.
In GFP curve it's possible to appreciate that in <partinfo>BBa_K300088</partinfo>, <partinfo>BBa_K300090</partinfo>, <partinfo>BBa_K300099</partinfo> GFP accumulation it's very similar and it's significantly different from that of negative control <partinfo>BBa_B0031</partinfo>. These results show the right folding of the green fluorescent protein assembled downstream of the genetic circuit.
The mean protein synthesis rate was also computed over the growth exponential phase, showing again an appreciable GFP production rate that is about a half of the positive control.
3OC6HSL inducible devices
Methods
Inoculum (into 5 ml LB+Amp) from glycerol stock of:
- <partinfo>BBa_K300091</partinfo>
- <partinfo>BBa_K300092</partinfo>
- <partinfo>BBa_K173000</partinfo> (positive control)
- <partinfo>BBa_B0031</partinfo> (negative control)
They were let grow ON at +37°C, 220 rpm.
The following day cultures were diluted 1:100 and let grow again for about five hours at +37°C, 220 rpm.
Optical density (O.D.) of each cell culture was than measured with TECAN Infinte F200. Samples were diluted in order to obtain the same O.D. equal to 0,02.
Than we performed a 21 hours' experiment with measurements of absorbance and green fluorescence every five minutes with TECAN Infinite F200. <partinfo>BBa_K300091</partinfo> and <partinfo>BBa_K300092</partinfo> circuits were induced 100nM with HSL directly into multiplate well. Each value shown is the mean of three measurements, from GFP data that of a non-fluorescent culture (negative control) was subtracted; cultures were shaken for 15 seconds every five minutes.
Results
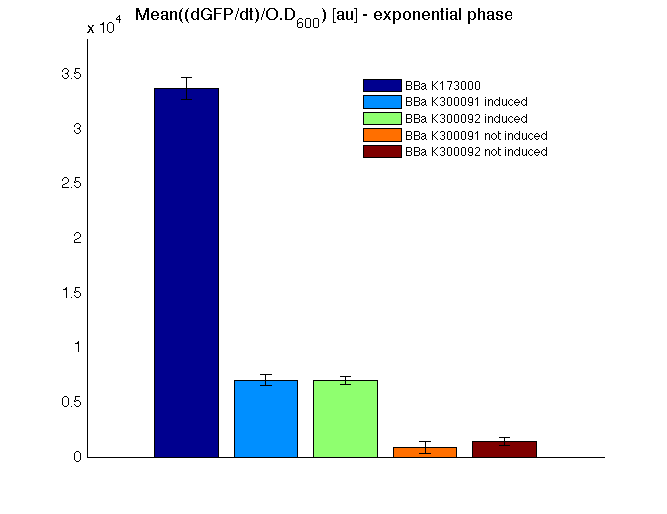 Mean (dGFP/dt)/O.D. over the exponential phase (under the hypothesis that GFP half-life in fusion contructs is similar to the original one - <partinfo>BBa_E0040</partinfo>) |
| Culture | Doubling time [min.] |
| <partinfo>BBa_K173000</partinfo> | 75 |
<partinfo>BBa_K300091</partinfo>
induced | 121 |
<partinfo>BBa_K300091</partinfo>
not induced | 74 |
<partinfo>BBa_K300092</partinfo>
induced | 123 |
<partinfo>BBa_K300092</partinfo>
not induced | 72 |
| <partinfo>BBa_B0031</partinfo> | 74 |
Discussion
All cell cultures showed a similar growth curve and doubling time was computed as described here in order to have informations about the burden due to synthesis of such fusion proteins. It's possible to see that all doubling time are very similar except for induced cultures. In this case doubling time is much higher than posite control and not induced cultures; so it's possible to assert that in this case there's a kind of metabolic burden higher than in the others, maybe because of the inducible system.
In GFP curve it's possible to appreciate that in induced <partinfo>BBa_K300091</partinfo> and <partinfo>BBa_K300092</partinfo> GFP accumulation profile it's very similar and it's significantly different from that of negative control <partinfo>BBa_B0031</partinfo>. On the other hand not induced <partinfo>BBa_K300091</partinfo> and <partinfo>BBa_K300092</partinfo> show a profile very similar to the last one. These results show the right folding of the green fluorescent protein assembled downstream of the genetic circuit and that the inducible system works as expected.
The mean protein synthesis rate was also computed over the growth exponential phase, showing again an GFP production rate that is different from negative control. Not induced <partinfo>BBa_K300091</partinfo> and <partinfo>BBa_K300092</partinfo> show a low GFP synthesis rate maybe due to 3OC6HSL inducible circuit leakage activity.
|
 "
"















