|
|
| (49 intermediate revisions not shown) |
| Line 26: |
Line 26: |
| | <!---------------------------------------PAGE CONTENT GOES BELOW THIS----------------------------------------> | | <!---------------------------------------PAGE CONTENT GOES BELOW THIS----------------------------------------> |
| | | | |
| - | =Using Recombineering to create a constitutive T6SS=
| |
| - | [[Image:Washington Recombineering.jpg|400px|thumb|right|Insertion of T7 promoters]]
| |
| - | The process for using homologous recombination to replace the T6SS native promoters is simple. We begin with a previously characterised fosmid (a large, single-copy plasmid based on the F' plasmid) which contains all the genes in our T6SS. Of particular interest is the ''fha1-tssA1'' intergenic region containing the promoters. We transform this fosmid into recombineering strain ''E. coli'' SW102, which contains heat-inducible lambda phage recombinases and lacks the galactose metabolism gene ''galK''.
| |
| | | | |
| - | Using PCR, we create a cassette containing the ''galK'' gene flanked by 50 bp of homology to the intergenic region. The lambda recombinases integrate the cassette into the fosmid. The transformants are plated on minimal galactose media to select for the recombinants.
| + | =Recombineering the Promoter Region of the Type VI Secretion System= |
| | + | [[Image:Washington_Recombineering_basic.png|750px|center|thumb|Schematic Showing the Engineering of the Fosmid to Contain a Divergent T7 Promoter Instead of the Native ''Pseudomonas'' Promoter]] |
| | + | Our goal was to replace the native ''P. aeruginosa'' regulation of the Type VI Secretion System with one that can be utilized by the ''E. coli'' transcriptional machinery. In order to replace the native promoters with more robust T7 promoters we used a technique called [https://2010.igem.org/Team:Washington/Protocols/Recombineering Recombineering] to flip out the region containing both promoters and replace it with a gene cassette as follows. We designed primers to create a cassette using PCR, containing the ''galK'' galactose metabolism gene flanked by 50 base pairs of homology just outside of the promoter region in the fosmid. The cassette was inserted, giving us a selection factor for our final insertion of our new promoters. A new promoter cassette was designed in total using oligos that encoded the two T7 promoters flanked by the same 50 base pairs of homology used previously, and inserted using recombineering into the fosmid that contained ''galK'' in the promoter region. |
| | | | |
| - | A second cassette is created via PCR, this time containing our new promoters. In this case, we chose to use T7 phage promoters due to their constitutive expression and small size, making them easier to synthesize. The promoters are again flanked with homologous regions, and integrated into the fosmid. This cassette displaces the ''galK'' cassette. The transformants are this time plated on 2-deoxy-galactose (DOG), which is a toxic homolog to galactose. Cells which can metabolize galactose (that is, those with a functioning ''galK'' gene) are killed, while those without the gene are unharmed.
| + | =Engineering the Toxin Antitoxin HSL-inducible circuit= |
| | + | [[Image:Washington_Building_Tse2_circuit.png|center|750px|thumb|Schematic Detailing the Transfer of Tse2 and Tsi2 from ''P. Aeruginosa'' into pSB3k3]] |
| | + | The Tse2/Tsi2 locus was amplified from ''P. aeruginosa'' genomic DNA using PCR. Primers were used that amplified the region from the start codon of Tse2 to the stop codon of Tsi2. The Tse2 and Tsi2 locus was then placed downstream from F2620 via [[Team:Washington/Tools_Used/Next-Gen_Cloning|Gibson Cloning]]. Gibson cloning is a method that joins regions of homology found on the 5' and 3' ends of linearized molecules. The end result of this Gibson cloning reaction is a circularized plasmid containing the final F2620.Tse2.Tsi2 construct in pSB3K3. The circularized plasmid is then transformed into electrocompetent ''E. coli'' Dh5a and the resulting colonies were screened for the insert of expected length via double restriction digest followed by agarose gel electrophoresis. The plasmids that showed inserts of expected length were sequenced, and it was determined that multiple plasmids had correct F2620.Tse2.Tsi2 inserts. |
| | | | |
| - | The final step is to transform the recombinant plasmid into a T7 polymerase expression strain, to allow T6SS constitutive expression.
| |
| | | | |
| - | =Increasing target specificity of a T6SS ''E. coli'' strain.=
| |
| - |
| |
| - | ==Why induce tse2 expression only when a gram-negative pathogen is present?==
| |
| - | A type six secretion system +/Tse2 +/Tsi2+ strain of ''E. coli'' could be administered like a traditional antibiotic after a pathogen infection by having a strain that constitutively expresses Tse2 and Tsi2. However, this would be a fairly brute force method of anti-bacterial comparable to traditional chemical antibiotics in its damage to the gut’s natural flora, and is only advantage over chemical antibiotics would be the current lack of Tse2 resistant pathogenic bacterial strains. If T se2 expression is induced by a chemical signal indicative of the presence of a pathogen, any unintended disruption of the natural gut flora will be limited to the area infected with pathogens, and the risk of Tse2 resistance would decrease. In addition, if Tse2 is only expressed when pathogenic bacteria are present, a Tse2+/ Tsi2+/ T6SS strain could be used in order to help prevent pathogenic gut infections, and could be a permanent member of the gut flora. Such a preventative strain could even be used against multiple gut pathogens by merely including multiple copies of tse2, each of which would respond to signals indicative of different pathogens.
| |
| - | ==3OC6HSL as a proxy for a pathogenic signal==
| |
| - | [[Image:3OC6HSL.gif|thumb|200px|Figure 1: Structure of 3OC6HSL|right]]
| |
| - | 3-oxohexanoyl-homoserine lactone (3OC6HSL) is a small organic molecule produced by ''Vibrio fischeri'' as a quorum sensing signal. Quorum sensing is a pathway where every bacterial cell produces and excretes a specific signalling molecule. When a specific density of bacteria is present in a region ( a quorum), the level of quorum sensing molecules is sufficient to induce or repress specific genes in order to tune gene expression to an environment of high cellular density. In ''V. fischeri'', 3OC6HSL permeates the cellular membrane and binds to the transcriptional factor LuxR, changing the conformation of LuxR in such a way that LuxR can bind to a DNA sequence within the Lux promoter, activating downstream transcription. While the 3OC6HSl-LuxR quorum sensing system is not present in any human pathogens, placing Tse2 downstream from a Lux promoter in a strain expressing LuxR does provide a good proof of concept showing that Tse2 can be regulated in such a way that Tse2 expression occurs only in the presence of a target bacterial pathogen( Figure 2). Many pathogenic bacteria do engage in quorum sensing. This system could be easily converted to express Tse2 only in the presence of a specific pathogenic species by changing the promoter and transcriptional factor. A major advantange of using the luxR quorum sensing system is that a well charactarized part containing both the LuxR transcriptional factor and Lux promoter already exists in the registry([http://partsregistry.org/Part:BBa_F2620 F2620]). This allowed for amplification from genomic DNA to be skipped, saving time.
| |
| - |
| |
| - | ==Plan to use a F2620-Tse2 plasmid in Tsi2- cells to measure toxicity of Tse2 in 'E. coli'==
| |
| - | Our initial plan was to tranform the F2620-Tse2 construct ( figure 2) into 'E. coli' cells not containing a copy of Tsi2. By increasing levels of 3OC6HSL in the culture medium of F2620-Tse2 construct + strains, one would expect to increase the amount of Tse2 transcription that would occur. By measuring relative cell growth in cultures containing increasing levels of 3OC6HSL ( this would be accomplished by measuring absorbance of liquid cultures at 600nm), one could get an indication of relative Tse2 level needed to cause 'E. coli' cell death. The toxicity of Tse2 could then be compared to that of other cellular toxins by replacing Tse2 in the F2620-Tse2 construct with other cellular toxins. Since culture 3OC6HSL levels is directly related to transcription from the Lux promoter, proteins that cause cell death only at higher 3OC6HSL are less toxic than proteins that cause cell death at lower 3OC6HSL concentrations. Characterization of toxicity of cell death proteins would allow for the correct choice of cell death protein for a given application.
| |
| - | [[image:tse2_F2620_circuit.png|frame||center|Figure 2: Diagram of F2620-Tse2 construct ]]
| |
| - | =Issues with building the F2620-Tse2 construct=
| |
| - | Tse2 was successfully amplified from ''Pseudomonas aeruginosa'' genomic DNA. However, it proved dificult to place Tse2 downstream from part F2620 to form the proposed F2620-Tse2 construct described above. All of the transformants that were sequenced showed mutations, either in Tse2 or in the pLux promoter. The fact that so many transformations showed mutations implies that the leakiness of the promoter causes enough tse2 production to cause a significant selection pressure against Tse2 production, implying that even small cellular levels of Tse2 is enough to cause cell death. One transformant displayed a deletion within pLux(figure 3), and it was decided that this transformation be tested for Tse2 production in the presence of 3OC6HSL, as it was posible that this mutation may reduce leakiness while still producing Tse2 in the presence of 3OC6HSL.
| |
| - | == phenotypic test of F2620-Tse2==
| |
| - | [[Image:Washington_test_growth_curve.png|400px|thumb|right|Figure 4: growth curve of F2620-Tse2]]
| |
| - | [[Image:Washington F2620 growth Control.png|400px|thumb|right|Figure 5: growth curve of F2620 control]]
| |
| - | If the F2620-Tse2 contruct was acting as expected, one would see cell death at high concentrations of 3OC6HSL, but cell survival when 3OC6HSL was not present. This test was conducted on MG1655 cultures containing F2620-Tse2 in pSB3k3, with cultures containing F2620 in pSB3k3 as a control. This test was conducted in a 96-well plate containing LB-kanamycin-3OC6HSL cultures. Each column of the plate contained increasing concentrations of 3OC6HSL ranging from 0M to 10,000 nM. 3OC6HSL was introduced into each plate via a concentrated 3OC6HSL-acidified ethyl acetate solution. The ethyl acetate was allowed to evaporate, leaving concentrated 3OC6HSL in the bottom of each well. LB broth and kanamycin was then added to each well. Each row was then innoculated with a small amount of a liquid overnight culture of either F2620-psB3k3 or F2620/Tse2-psB3k3 so that the final 600nm optical density was approximetly .01. Both the F2620-psB3k3 and F2620/Tse2-psB3k3 cultures were tested in triplicate at each concentration of 3OC6HSL, and one row was used as a blank measurement. The cultures were then grown in a plate reader at 37°C. Optical density readings at 600nm were taken every 5 minutes for a period of approximently 21 hours. The F2620-Tse2 cultures (figure 4) showed no significant difference in growth from the F2620 control cultures( figure 5) at any concentration of 3OC6HSL. This is consistent with the promoter mutation resulting in non-transcription of Tse2.
| |
| - | == Western blotting of F2620-Tse2 cultures==
| |
| - | In order to confirm that Tse2 was not being produced, SDS-PAGE followed by western blotting wwas performed on pellets from DH5a and MG1655 overnight cultures grown in conditions either with or without 3OC6HSL. No culture showed a positive western blot, implying that no Tse2 is being produced, either by leakiness or proper production due to the presence of 3OC6HSL.
| |
| - |
| |
| - | [[Image:Washington_test_growth_curve.png|400px|thumb|right|Figure 4: growth curve of F2620-Tse2]]
| |
| | <!---------------------------------------PAGE CONTENT GOES ABOVE THIS----------------------------------------> | | <!---------------------------------------PAGE CONTENT GOES ABOVE THIS----------------------------------------> |
| | | | |
| | <div style="text-align:center"> | | <div style="text-align:center"> |
| - | '''← [[Team:Washington/Project/Mougous/Design|Designing the Gram(-) Therapeutic]]''' | + | '''← [[Team:Washington/Gram Negative/Design|Designing the Gram(-) Therapeutic]]''' |
| | | | |
| | | | |
| | | | |
| - | '''[[Team:Washington/Project/Mougous/Test|Testing the Gram(-) Therapeutic]] →''' | + | '''[[Team:Washington/Gram Negative/Test|Testing the Gram(-) Therapeutic]] →''' |
| | </div> | | </div> |
| | | | |
| | {{Template:Team:Washington/Templates/Footer}} | | {{Template:Team:Washington/Templates/Footer}} |
Recombineering the Promoter Region of the Type VI Secretion System
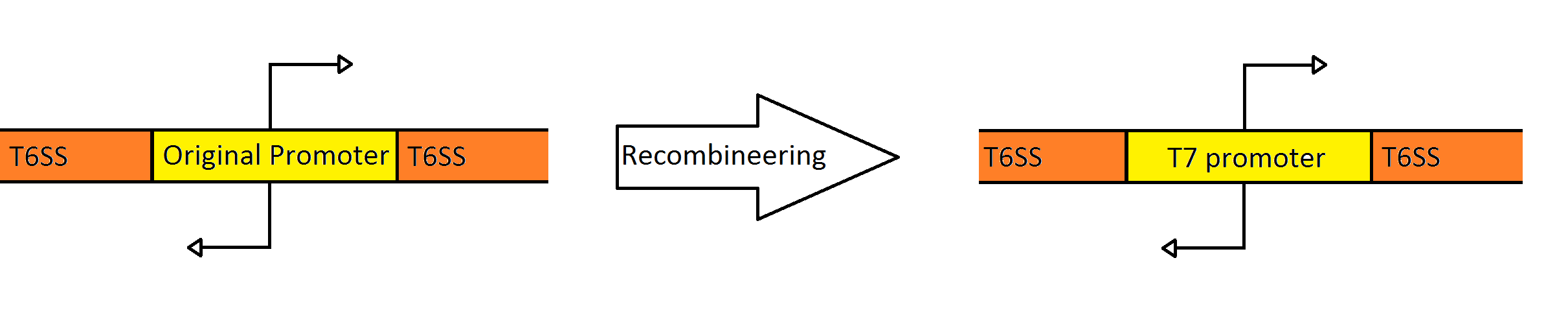
Schematic Showing the Engineering of the Fosmid to Contain a Divergent T7 Promoter Instead of the Native
Pseudomonas Promoter
Our goal was to replace the native P. aeruginosa regulation of the Type VI Secretion System with one that can be utilized by the E. coli transcriptional machinery. In order to replace the native promoters with more robust T7 promoters we used a technique called Recombineering to flip out the region containing both promoters and replace it with a gene cassette as follows. We designed primers to create a cassette using PCR, containing the galK galactose metabolism gene flanked by 50 base pairs of homology just outside of the promoter region in the fosmid. The cassette was inserted, giving us a selection factor for our final insertion of our new promoters. A new promoter cassette was designed in total using oligos that encoded the two T7 promoters flanked by the same 50 base pairs of homology used previously, and inserted using recombineering into the fosmid that contained galK in the promoter region.
Engineering the Toxin Antitoxin HSL-inducible circuit
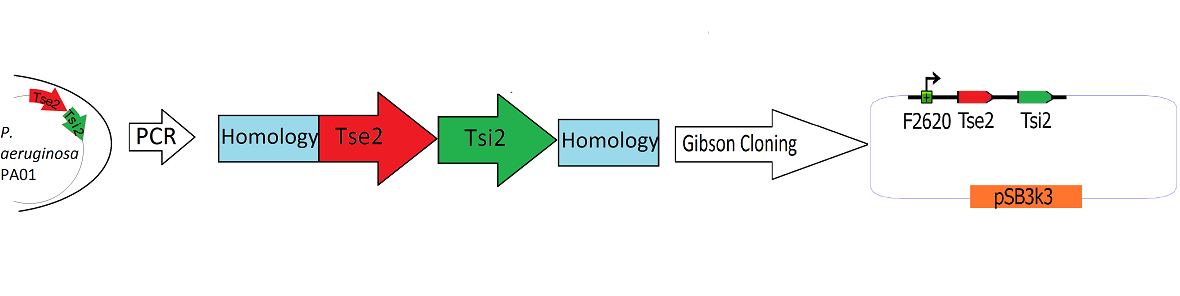
Schematic Detailing the Transfer of Tse2 and Tsi2 from
P. Aeruginosa into pSB3k3
The Tse2/Tsi2 locus was amplified from P. aeruginosa genomic DNA using PCR. Primers were used that amplified the region from the start codon of Tse2 to the stop codon of Tsi2. The Tse2 and Tsi2 locus was then placed downstream from F2620 via Gibson Cloning. Gibson cloning is a method that joins regions of homology found on the 5' and 3' ends of linearized molecules. The end result of this Gibson cloning reaction is a circularized plasmid containing the final F2620.Tse2.Tsi2 construct in pSB3K3. The circularized plasmid is then transformed into electrocompetent E. coli Dh5a and the resulting colonies were screened for the insert of expected length via double restriction digest followed by agarose gel electrophoresis. The plasmids that showed inserts of expected length were sequenced, and it was determined that multiple plasmids had correct F2620.Tse2.Tsi2 inserts.
 "
"


