 "
"
Team:UIUC-Illinois/Project/Protocols
From 2010.igem.org
(→Protocols) |
(→Protocols) |
||
| (4 intermediate revisions not shown) | |||
| Line 3: | Line 3: | ||
== Protocols == | == Protocols == | ||
| + | |||
| + | <h3>Digestions</h3> | ||
| + | Reaction Mix: | ||
| + | * 500 ng – Template | ||
| + | * 5 µl – 10x Buffer | ||
| + | * 1 µl – NEB Enzyme 1 (10 Units/µl) = 10 Units in 50 µl reaction | ||
| + | * 1 µl – NEB Enzyme 2 (10 Units/µl) = 10 Units in 50 µl reaction | ||
| + | * dH2O to 20 µl | ||
| + | TOTAL – 50 µl | ||
| + | Procedure: | ||
| + | # Calculate how much template is needed for digestion. Then calculate how much water needs to be added to make a 50 µl reaction. | ||
| + | # Pipette appropriate amounts of dH20, template, and 10x Buffer in that order into a PCR tube (200 µl). Mix reagents together by vortexing and then tapping tube on desk to keep reagents on the bottom of the PCR tube. | ||
| + | # Add appropriate amounts of Enzyme 1 and Enzyme 2. Once added gently swirl around reaction mix with pipette tip. | ||
| + | # Incubate at 37o C for 1 – 2 hours | ||
| + | # Run a gel of your digestions on a gel with low temperature melting agarose. Run at 100V for 1 hr. 30 min. | ||
| + | # Look at gel under 320nm UV light. Conduct gel extraction purification. | ||
| + | NOTE: When digesting your vector it may be useful to use 2 µl of each enzyme in the 50 µl reaction and extending incubation time. | ||
| + | |||
| + | |||
| + | <h3>Electroporation (Transformation)</h3> | ||
| + | Supplies: | ||
| + | * 100 µl – Electrocompetent Cells | ||
| + | * 2-5 µl – Ligation Reaction (10 – 100 ng DNA) | ||
| + | * 1 ml – SOC broth | ||
| + | * Electrocuvette | ||
| + | Procedure: | ||
| + | # Gather all your supplies and keep them on ice (or in 4o C room). | ||
| + | # Thaw frozen electrocompetent cells on ice and place in electrocuvette. Then add ligation reaction and tap the cuvette to mix well. DO NOT pipette up and down. | ||
| + | # Keep on ice for 1 min. | ||
| + | # Pulse once (On machine use Ec1 for 1mm cuvettes and Ec2 for 2mm cuvettes). | ||
| + | # IMMEDIATELY after add 1ml of SOC broth and pipette up and down gently. | ||
| + | # Incubate recovering cells in 37o C for 1 hr. | ||
| + | Plating | ||
| + | * After incubation, plate 200 µl of transformants on to appropriate selective plate and incubate at 37o C for 18+ hrs. | ||
| + | * If no colonies are formed plate out the rest of the transformants (~800 µl) and incubate at 37o C for 24 hrs. | ||
| + | * Pray it’ll work. | ||
| + | Heat Shock Transformation | ||
| + | Supplies: | ||
| + | * 50 µl – Chemically Competent Cells | ||
| + | * 2-5 µl – Ligation Reaction (10 – 100 ng DNA) | ||
| + | * 1 ml – SOC broth | ||
| + | Procedure: | ||
| + | # Gather supplies, be sure to keep cells on ice | ||
| + | # Thaw competent cells on ice for 5 minutes | ||
| + | # Add 2-5 µl of ligation reaction to cells, swirl with pipette tip, and keep on ice for 2 minutes | ||
| + | # Place transformation reaction in a 42𝑜 C water bath for 45 seconds | ||
| + | # After heating put reaction on ice for 10 min | ||
| + | # Add 1 mL of SOC broth and incubate at 37o C for 1 hr | ||
| + | # Plate | ||
| + | |||
<h3>Ligations</h3> | <h3>Ligations</h3> | ||
| Line 9: | Line 59: | ||
# Estimate the intensities of your insert and vector where your insert (usually less intense) is considered 1 and your vector is x times as bright. | # Estimate the intensities of your insert and vector where your insert (usually less intense) is considered 1 and your vector is x times as bright. | ||
# Once intensity is determined perform the following equation: | # Once intensity is determined perform the following equation: | ||
| + | #* [[Image:Lunapic 128814558288331 1.png]] | ||
#* L = Ligation volume S = Size of fragments I = Intensity of fragments V = Volume ran down gel i = Insert v = Vector | #* L = Ligation volume S = Size of fragments I = Intensity of fragments V = Volume ran down gel i = Insert v = Vector | ||
# Ligation volume (Lv) for the vector should be approximately 2 µl. | # Ligation volume (Lv) for the vector should be approximately 2 µl. | ||
Latest revision as of 02:17, 27 October 2010

Contents |
Protocols
Digestions
Reaction Mix:
- 500 ng – Template
- 5 µl – 10x Buffer
- 1 µl – NEB Enzyme 1 (10 Units/µl) = 10 Units in 50 µl reaction
- 1 µl – NEB Enzyme 2 (10 Units/µl) = 10 Units in 50 µl reaction
- dH2O to 20 µl
TOTAL – 50 µl Procedure:
- Calculate how much template is needed for digestion. Then calculate how much water needs to be added to make a 50 µl reaction.
- Pipette appropriate amounts of dH20, template, and 10x Buffer in that order into a PCR tube (200 µl). Mix reagents together by vortexing and then tapping tube on desk to keep reagents on the bottom of the PCR tube.
- Add appropriate amounts of Enzyme 1 and Enzyme 2. Once added gently swirl around reaction mix with pipette tip.
- Incubate at 37o C for 1 – 2 hours
- Run a gel of your digestions on a gel with low temperature melting agarose. Run at 100V for 1 hr. 30 min.
- Look at gel under 320nm UV light. Conduct gel extraction purification.
NOTE: When digesting your vector it may be useful to use 2 µl of each enzyme in the 50 µl reaction and extending incubation time.
Electroporation (Transformation)
Supplies:
- 100 µl – Electrocompetent Cells
- 2-5 µl – Ligation Reaction (10 – 100 ng DNA)
- 1 ml – SOC broth
- Electrocuvette
Procedure:
- Gather all your supplies and keep them on ice (or in 4o C room).
- Thaw frozen electrocompetent cells on ice and place in electrocuvette. Then add ligation reaction and tap the cuvette to mix well. DO NOT pipette up and down.
- Keep on ice for 1 min.
- Pulse once (On machine use Ec1 for 1mm cuvettes and Ec2 for 2mm cuvettes).
- IMMEDIATELY after add 1ml of SOC broth and pipette up and down gently.
- Incubate recovering cells in 37o C for 1 hr.
Plating
- After incubation, plate 200 µl of transformants on to appropriate selective plate and incubate at 37o C for 18+ hrs.
- If no colonies are formed plate out the rest of the transformants (~800 µl) and incubate at 37o C for 24 hrs.
- Pray it’ll work.
Heat Shock Transformation Supplies:
- 50 µl – Chemically Competent Cells
- 2-5 µl – Ligation Reaction (10 – 100 ng DNA)
- 1 ml – SOC broth
Procedure:
- Gather supplies, be sure to keep cells on ice
- Thaw competent cells on ice for 5 minutes
- Add 2-5 µl of ligation reaction to cells, swirl with pipette tip, and keep on ice for 2 minutes
- Place transformation reaction in a 42𝑜 C water bath for 45 seconds
- After heating put reaction on ice for 10 min
- Add 1 mL of SOC broth and incubate at 37o C for 1 hr
- Plate
Ligations
Set-up:
- Run a gel of your digestions and take a picture.
- Estimate the intensities of your insert and vector where your insert (usually less intense) is considered 1 and your vector is x times as bright.
- Once intensity is determined perform the following equation:
-
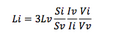
- L = Ligation volume S = Size of fragments I = Intensity of fragments V = Volume ran down gel i = Insert v = Vector
-
- Ligation volume (Lv) for the vector should be approximately 2 µl.
- The ratio for the ligation in the previous equation is 3:1 insert:vector but can be done 6:1 by replacing the 3 with a 6.
Reaction Mix:
• 2 µl – Vector
• Li µl – Insert
• 2 µl – 10x Buffer
• dH20 to 19 µl
• 1 µl – T4 DNA Ligase
TOTAL – 20 µl
Procedure:
- Add appropriate amounts of dH20, Vector, and Insert in a PCR tube (200 µl). Once added, heat mixture to 65o C for 5 min. This will remove all items that are already annealed to each other causing inefficient ligation.
- Add then appropriate amounts of 10x Buffer and T4 DNA Ligase. Make sure not to pipette up and down or vortex to mix, this will shear the ligase. Gently swirl with the tip of your pipette to mix.
- Incubate reaction at room temperature for 30 minutes.
- After incubation heat inactivate the ligase by another incubation at 80o C for 20 min.
NOTE: The ligase buffer should smell like wet dog. If you smell nothing from the tube, then the buffer is old and useless.
Making Electrocompetent Cells
SOB
2% tryptone
0.5% yeast extract
10 mM NaCl
2.5 mM KCl
10 mM MgCl2
10 mM MgSO4
SOC
SOB + 20 mM glucose
Sterile 10% glycerol (can be autoclaved) is needed for the washes. The volume of 10% glycerol needed is 2X the culture volume (for example, a 500 ml culture requires 1L of 10% glycerol).
Procedure (for 2, 250 ml cultures)
- Inoculate 1 colony from a fresh plate of the strain to be made electrocompetent into 10 ml of SOB in a 125 ml flask and incubate for 16-18 hours at 37oC and 250 rpm.
- Have ready 2, 1 L flasks containing 250 ml each of SOB pre-warmed to 37°C. Add two drops of the overnight culture to each of the flasks.
- Shake at 37°C and 250 rpm until the cultures reach an OD600 of 0.5-0.7. Be sure to turn on centrifuge and cool rotor to 4°C well in advance of harvesting cells. Be sure to place 1 L of 10% glycerol on ice well in advance of harvesting cells
- Place cultures on ice for 15 minutes. From this point on the cultures must be kept ice cold. Pour each 250 ml culture into chilled 500 ml (or 1000 ml) centrifuge bottles.
- Centrifuge at 5000 rpm for 10 min. Pour off the supernatant and aspirate any residual broth.
- Add 250 ml of glycerol to each of the centrifuge bottles and completely suspend the cells by pipetting up and down.
- Centrifuge at 5000 rpm for 10 min. Pour off the supernatant, it is not necessary to aspirate. Completely suspend the cells as before.
- Pour off the supernatant and suspend the cells in the residual glycerol by pipetting up and down.
- At this point you can electroporate or freeze the cells away. To freeze, Add 100 microliters of the culture to microcentrifuge tubes on ice. Once you have used all of the culture transfer the tubes to dry ice for 10 minutes. Once the cultures are frozen transfer them to a -80°C freezer. The cultures should be good for >6 months.
Site Directed Mutagenesis
Procedure modified from [http://www.genomics.agilent.com/files/Manual/200555B_01.pdf Stratagene QuikChange II Protocol]
Materials
- PFU Ultra Polymerase (high fidelity)
- 10X Reaction Buffer
- DpnI (20U/μL)
- dNTPs
- Competent Cells
1. Design primers
2. Mutant strand synthesis (PCR)
3. DpnI digestion of template
4. Transform and plate
Primer Design Considerations
• Both primers must contain desired mutation and anneal to same sequence on opposite strands of the plasmid.
• Keep the Primers aruond 25-45 bp long with 10-15 bp on either side of the mutated base.
• Keep the melting temperature greater than or equal to 78°C
Tm=81.5+,41(%GC)-675/N-%mismatch where N is the length of the primer
• Try to keep the primers to a minimum GC content of 40% and end the primer in GC for a clamp.
• Make sure primers are in excess to template
PCR
| Ingredient | Amount |
| 10X Buffer | 5 μL |
| Template | 20 ng |
| Primer 1 | 125 ng |
| Primer 2 | 125 ng |
| 10 mM dNTPs | 1 μL |
| Polymerase | 1 μL |
| H2O | Bring to 50 μL |
| 95°C | 3 min |
| 95°C | 30 sec |
| annealing temp | 1 min |
| 72°C | 1 min/Kb |
| go to step 2 | 12X |
| 72°C | 1 min | hold at 4°C |
DpnI Digestion
| DNA | 500 ng |
| Buffer 4 | 5 μL |
| BSA | .5 μL |
| DpnI | 1 μL |
| H2O | Bring to 50 μL |
Transform via electroporation