
Self-inducible promoters

Integrative standard vector for E. coli
The integration of the genetic circuits of interest into the microbial host genome can eliminate the need of expensive selection techniques, such as antibiotics or auxotrophic media, in cell cultures.
In order to simplify the engineering of the host genome, two standard and modular integrative vectors have been designed for Escherichia coli and Saccharomyces cerevisiae, two commonly used hosts for industrial protein production. Here, a detailed description of the integrative vector for E. coli is reported, while the following section deals with the integrative vector for yeast.
The structure of the designed vector, here named <partinfo>BBa_K300000</partinfo>, is shown in Fig.1. Most of its features have been inspired by <partinfo>BBa_I51020</partinfo> (BioBrick base vector) and <partinfo>BBa_J72007</partinfo> (BamHI methyltransferase encoding CRIM plasmid), described by [Shetty RP et al., 2008] and [Anderson JC et al., 2010] respectively.
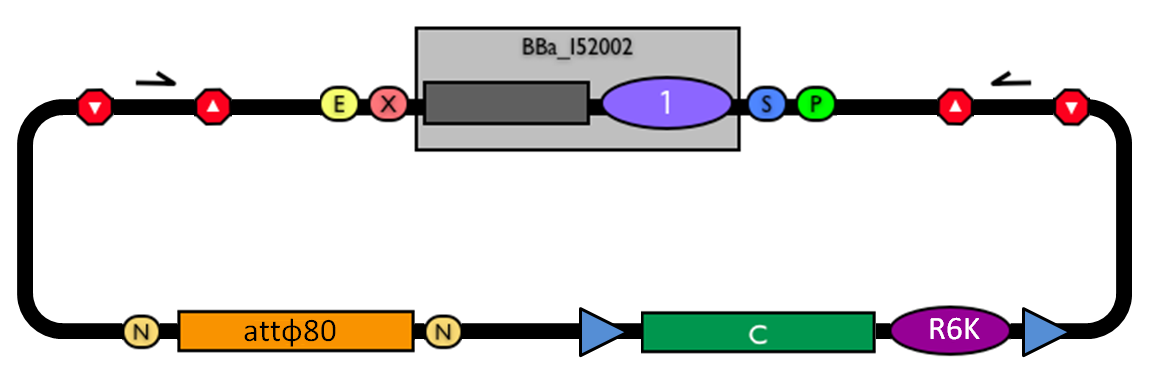 Figure 1: BioBrick integrative base vector BBa_K300000. | 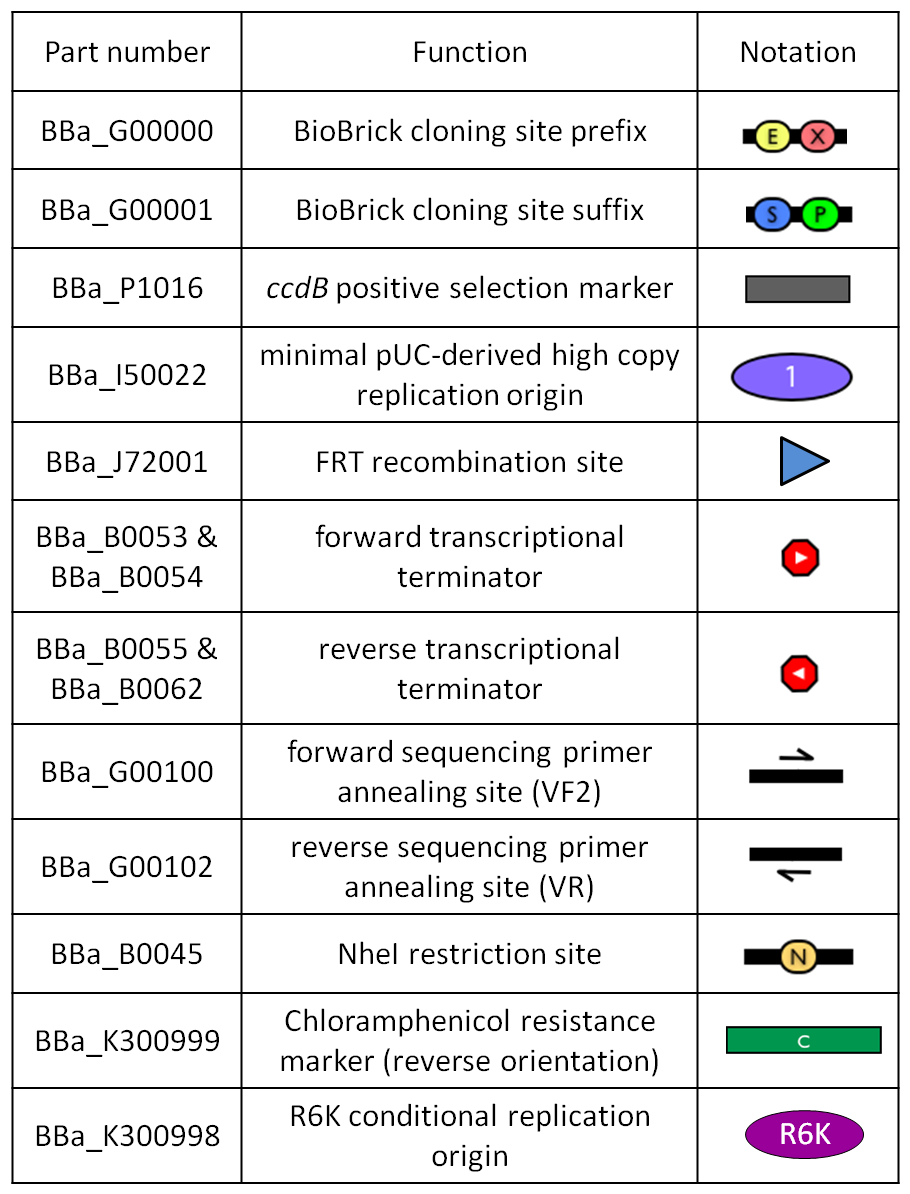 Figure 2: Parts notation. |
This vector can be considered as a base vector, which can be specialized to target the desired integration site in the host genome. The default version of this backbone has the bacteriophage Phi80 attP (<partinfo>BBa_K300991</partinfo>) as integration site.
This vector enables multiple integrations in different positions of the same genome.
Glossary
The passenger is the desired DNA part to be integrated into the genome.
The guide is the DNA sequence that is used to target the passenger into a specific locus in the genome.
The main design features for vector engineering and for the genome integration of the vector are reported below.
Vector engineering features:
- The cloning site is compatible with the original BioBrick standard (RFC10), i.e. it is composed by the BioBrick Prefix (<partinfo>BBa_G00000</partinfo>) and Suffix (<partinfo>BBa_G00001</partinfo>). The presence of illegal restriction sites (XbaI in <partinfo>BBa_J72001</partinfo> and SpeI in <partinfo>BBa_K300991</partinfo>) prevents the usage of this backbone in the classic BioBrick Standard Assembly process. However, the presence of unique EcoRI and PstI sites in Prefix and Suffix fully supports the assembly of the desired BioBrick parts in the cloning site upon EcoRI-PstI digestion and also supports the 3A Assembly.
- The two NheI restriction sites flanking the default integration guide sequence <partinfo>BBa_K300991</partinfo> enable the engineering of this backbone by assembling new user-defined BioBrick integration guides upon XbaI-SpeI digestion, if the desired guide conforms to the RFC10 or a compatible standard.
- The default insert <partinfo>BBa_I52002</partinfo> contains the positive selection marker <partinfo>BBa_P1016</partinfo> and the pUC19-derived replication origin <partinfo>BBa_I50022</partinfo>. <partinfo>BBa_P1016</partinfo> expresses the ccdB toxin gene, which is lethal for most E. coli strains and is useful to prevent the growth of transformants containing the uncut plasmid contaminant DNA. For this reason, the default vector must be propagated in ccdB-tolerant strains, such as DB3.1 (<partinfo>BBa_V1005</partinfo>). <partinfo>BBa_I50022</partinfo> enables the propagation of this vector at high copy in the used ccdB-tolerant strain.
- Like in many other standard vector backbones (e.g. the pSB**5 series), the binding sites for standard primers VF2 (<partinfo>BBa_G00100</partinfo>) and VR (<partinfo>BBa_G00102</partinfo>) are present upstream and downstream of the BioBrick cloning site respectively. These two sequences are sufficiently distant from the cloning site to enable a good quality sequencing of the insert.
Genome integration features:
- The four transcriptional terminators <partinfo>BBa_B0053</partinfo>, <partinfo>BBa_B0054</partinfo>, <partinfo>BBa_B0055</partinfo> and <partinfo>BBa_B0062</partinfo> ensure the transcriptional insulation of the integrated part from its flanking genome sequences.
- The two FRT recombination sites (<partinfo>BBa_J72001</partinfo>) enable the excision of <partinfo>BBa_K300994</partinfo>-<partinfo>BBa_K300998</partinfo>-<partinfo>BBa_G0001</partinfo>-<partinfo>BBa_B0025</partinfo>-<partinfo>BBa_G0001</partinfo>-<partinfo>BBa_K300999</partinfo>-<partinfo>BBa_K300995</partinfo> (i.e. the R6K origin and the Chloramphenicol resistance marker) upon Flp recombinase activity. This marker excision allows users to make multiple integrations in the same strain, always using the same antibiotic resistance marker.
- The engineering of the integration guide allows the integration of parts in user-defined genome positions and for this reason this vector supports the integration by exploiting bacteriophage attP-mediated integration as well as homologous recombination.
How to use it
<partinfo>BBa_K300000</partinfo> can be:
- propagated in E. coli
- engineered to change the passenger and/or the integration guide
- integrated into the desired locus of the host genome
- used to perform the desired number of serial integrations in the same genome
How to propagate it before performing genome integration
The default version of this vector contains the <partinfo>BBa_I52002</partinfo> insert, so it *must* be propagated in a ccdB-tolerant strain such as DB3.1 (<partinfo>BBa_V1005</partinfo>).
After the insertion of the desired BioBrick part in the cloning site, this vector does not contain a standard replication origin anymore, so it *must* be propagated in a pir+ or pir-116 strain such as BW25141 (<partinfo>BBa_K300984</partinfo>) or BW23474 (<partinfo>BBa_K300985</partinfo>) that can replicate the R6K conditional origin (<partinfo>BBa_J61001</partinfo>).
How to engineer it
The DNA guide can be changed as follows:
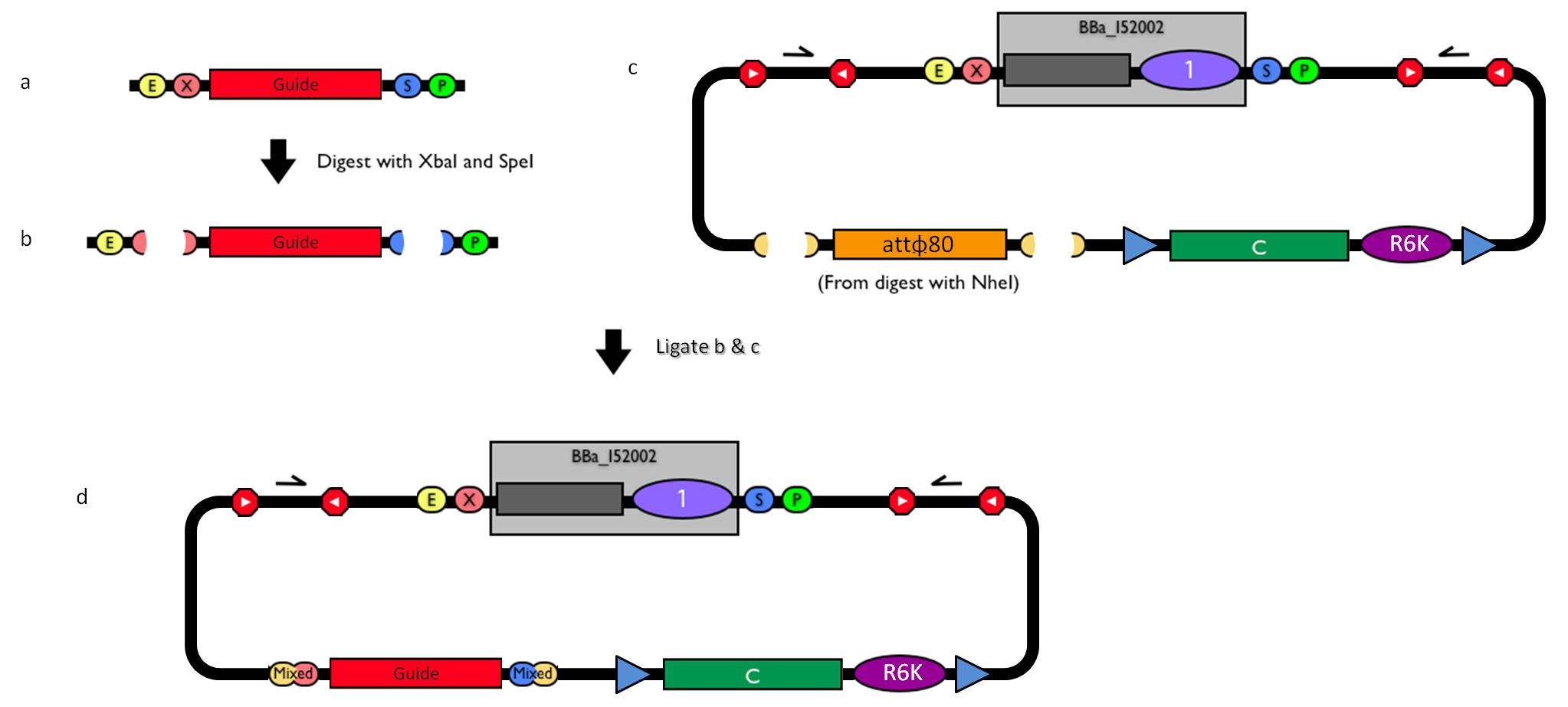 Figure 3: How to engineer the integrative base vector to assemble the desired DNA guide. |
- Be sure to have the desired guide in the RFC10 standard or a compatible one (Fig.3-a).
- Digest the guide with XbaI-SpeI (Fig.3-b).
- Digest the integrative base vector <partinfo>BBa_K300000</partinfo> with NheI (Fig.3-c) and dephosphorylate the linearized vector to prevent re-ligation.
- Ligate the digestion products (Fig.3-d). XbaI, SpeI and NheI all have compatible protruding ends. Note that the ligation is not directional, but the guide can work in both directions.
- Transform the ligation in a ccdB-tolerant strain and screen the clone.
The DNA passenger can be changed as follows:
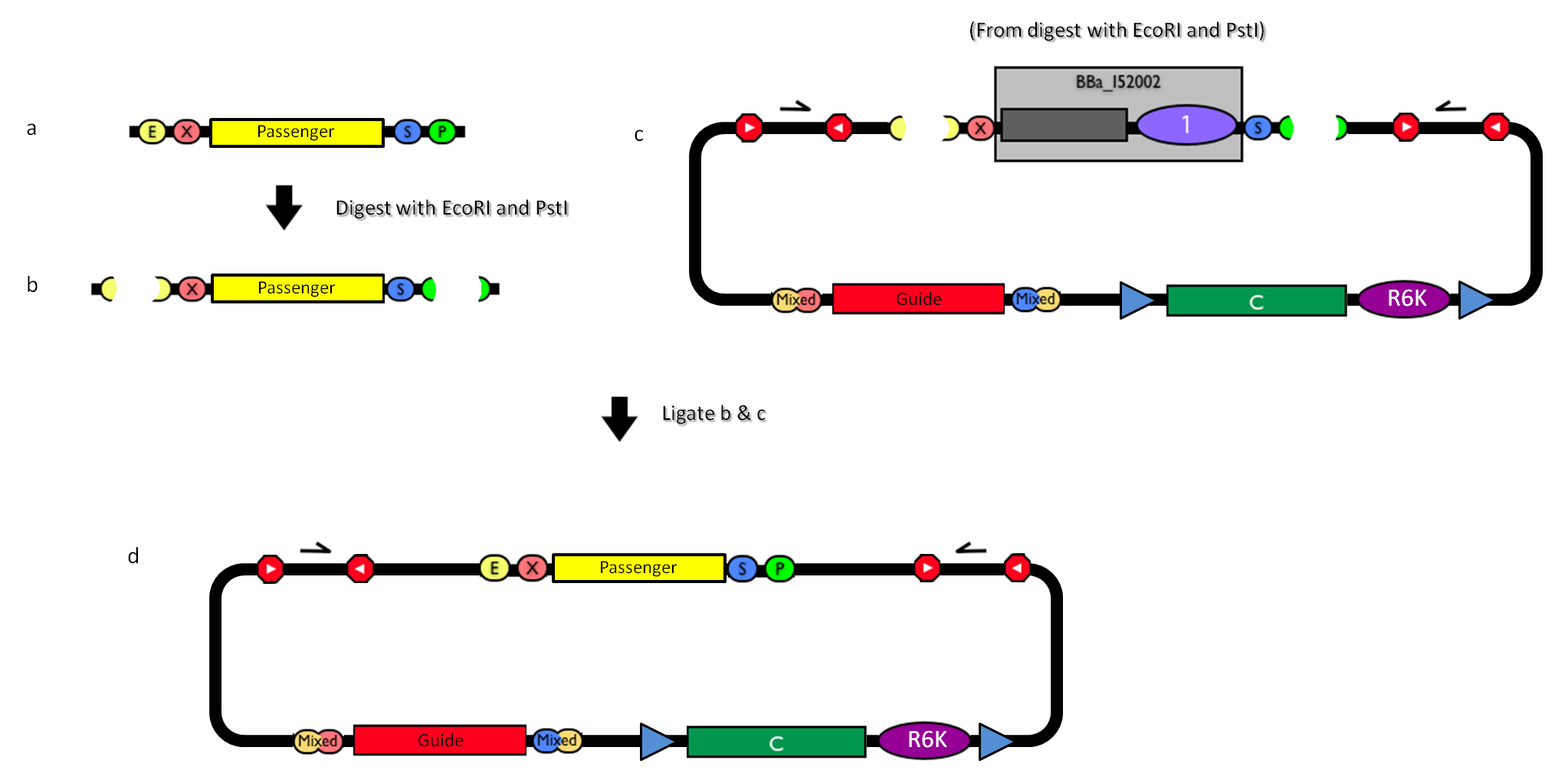 Figure 4: How to engineer the integrative base vector to assemble the desired DNA passenger. |
- Be sure to have the desired passenger in the RFC10 standard or a compatible one (Fig.4-a).
- Digest the passenger with EcoRI-PstI (Fig.4-b).
- Digest the integrative base vector <partinfo>BBa_K300000</partinfo> with EcoRI-PstI (Fig.4-c).
- Ligate the digestion products (Fig.4-d).
- Transform the ligation in a pir+/pir-116 strain. Transformants with the uncut plasmid contaminant DNA do not grow because of the ccdB toxin in <partinfo>BBa_I52002</partinfo>. Screen the clone.
How to perform genome integration
The integration into the E. coli chromosome can exploit the bacteriophage attP-mediated integration or the homologous recombination.
Detailed protocols about attP-mediated integration can be found here:
- Anderson JC et al., 2010
- Haldimann A and Wanner BL, 2001
Detailed protocols about homologous recombination can be found here:
- Martinez-Morales F et al., 1999
- Posfai G et al., 1997
When using the default integration guide <partinfo>BBa_K300991</partinfo>, the integration method relies on the bacteriophage site-specific recombination (attP-mediated recombination) through the attP site on the integrative vector and the attB site in the host genome.
This integration method is applicable when the host strain does not have prophages in the att(Phi80) locus. TOP10 (<partinfo>BBa_V1009</partinfo>) and DH5alpha (<partinfo>BBa_V1001</partinfo>) strains have the Phi80 prophage and so their chromosome cannot be engineered with this procedure.
The genomic integration of the desired BioBrick part into the attP(Phi80) locus has to be mediated by co-transforming a helper plasmid, such as the Amp-resistant <partinfo>BBa_J72008</partinfo> plasmid) which carries the IntPhi80 site-specific integrase gene under the control of a thermoinducible promoter (see Fig.5). The helper plasmid also has a heat-sensitive replication origin, whose replication can be inhibited at temperatures of 37-42°C, while a permissive temperature for this vector is 30°C. For this reason, it can be cured at high temperatures, when the integrase expression is triggered at the same time.
The Phi80 integrase mediates the site-specific recombination between the attP site in the integrative vector and the attB site in the bacterial genome (for a schematic description of this process, see Fig.6 and http://partsregistry.org/Recombination).
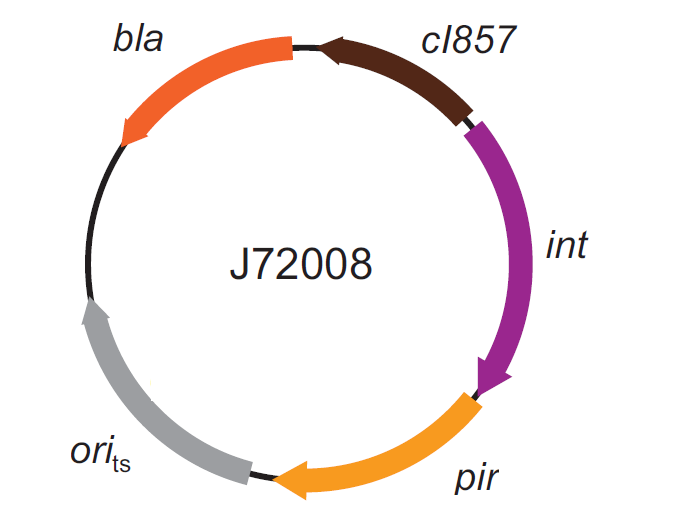 Figure 5: Schematic description of the <partinfo>BBa_J72008</partinfo> plasmid. cI857 is the expression system for the thermoinducible cI repressor; int is the Phi80 integrase regulated by the lambda cI-repressible promoter; pir is the expression system for the pir-116 gene which is able to trigger the propagation of the R6K conditional replication origin; ori_ts is the heat-sensitive replication origin (low copy) of the vector; bla is the Ampicillin resistance marker. | 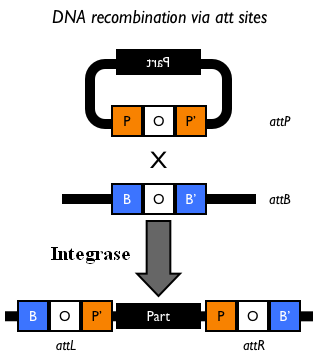 Figure 6: Schematic description of site-specific recombination between a bacteriophage attP attachment site in the plasmid and an attB attachment site in the bacterial genome. This process is mediated by a specific integrase. |
Thanks to its R6K conditional replication origin, the integrative vector cannot be replicated in common E. coli strains, so the Chloramphenicol resistant bacteria are actual integrants.
In the Materials and Methods section (https://2010.igem.org/Team:UNIPV-Pavia/Project/results), a detailed protocol to target the desired BioBrick part into the Phi80 locus is reported.
How to perform multiple integrations in the same genome
When this vector is integrated into the genome, the desired passenger should be maintained into the host, as well as the Chloramphenicol resistance marker and the R6K conditional replication origin. The CmR and the R6K can be excised from the genome by exploiting the two FRT recombination sites that flank them. The Flp recombinase protein mediates this recombination event, so it has to be expressed by a helper plasmid, such as pCP20 (CGSC#7629).
This enables the sequential integration of several parts using the same antibiotic resistance marker, which can be eliminated each time.
Detailed protocols about homologous recombination can be found here:
- Cherepanov PP and Wackernagel W, 1995
- Datsenko KA and Wanner BL, 2000
Materials and Methods
Plasmids and strains: the <partinfo>BBa_J72008</partinfo> helper plasmid was kindly given by Prof. JC Anderson (UC Berkeley). MC1061 (<partinfo>BBa_K300078</partinfo>) and MG1655 (<partinfo>BBa_V1000</partinfo>) E. coli strains and the pCP20 helper plasmid were purchased from the Coli Genetic Stock Center (Yale University). DH5alpha (<partinfo>BBa_V1001</partinfo>) strain was purchased from Invitrogen.
Verification primers: all the oligonucleotides were purchased from Primm (San Raffaele Biomedical Science Park, Milan, Italy). The P1 (<partinfo>BBa_K300975</partinfo>) and P4 (<partinfo>BBa_K300978</partinfo>) primers had already been used in [Anderson JC et al., 2010]. The P2 (<partinfo>BBa_K300976</partinfo>) and P3 (<partinfo>BBa_K300977</partinfo>) primers have been newly designed using ApE and amplifiX. P2 and P3 have been designed also considering the previously used verification primers P2 and P3 in the pG80ko integrative plasmid, described in [DeLoache W, 2009].
The relative position of the P1, P2, P3 and P4 primers is shown in the figure below:
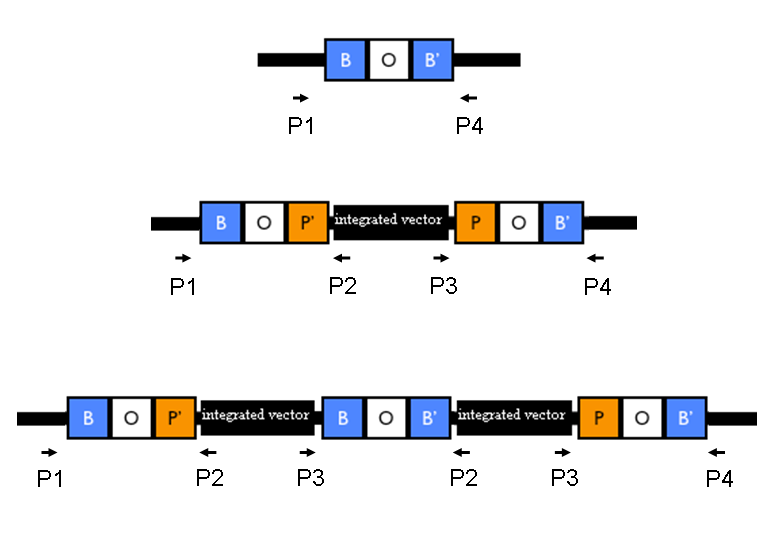 Relative position of the verification primers. a) no integrants; b) single integrant and c) integrant with multiple tandem copies. P1/P2 and P3/P4 pairs give an amplicon when at least one copy of the vector is integrated in the Phi80 locus. P2/P3 pair show an amplicon only when multiple tandem copies occur. |
Competent cells preparation: all the E. coli strains were made competent following a slightly modified version of the protocol described in [Sambrook J et al., 1989]. Briefly, cells were grown to and OD600 of ~0.4-0.6, harvested (4000 rpm, 10 min, 4°C) and the supernatant discarded. Cells were resuspended in (30 ml for each 50 ml of initial culture) pre-chilled Mg-Ca buffer (80 mM MgCl2, 20 mM CaCl2), centrifuged as before and the supernatant discarded. Cells were resuspended in (2 ml for each 50 ml of initial culture) pre-chilled Ca buffer (100 mM CaCl2, 15% glycerol), aliquoted in 0.5 ml tubes and freezed immediately at -80°C. Test the transformation efficiency as:
efficiency [CFU/ug of DNA]= # CFU * 1000 ng of DNA / amount of transformed DNA [ng]
Pir strains validation: in order to test the capability of BW25141 (pir+) and BW23474 (pir-116) to propagate this conditional replication origin, they were made competent, as well as three control strains: MC1061, MG1655 and DH5alpha. Then, a vial of 100 ul of competent cells was transformed with 2-4 ng of:
- no DNA (negative control);
- a pSB*** series vector (positive control);
- self-ligated <partinfo>BBa_K300008</partinfo> (a R6K plasmid with Cm resistance).
Self-ligated <partinfo>BBa_K300008</partinfo> was prepared by digesting <partinfo>pSB1A2</partinfo>-<partinfo>BBa_K300008</partinfo> (yielded by BioBrick Standard Assembly) with XbaI-SpeI. The insert was isolated and purified from a 1% agarose gel. Then, it was self-ligated to generate a Cm-resistant R6K plasmid. The colonies were counted in each plate and the transformation efficiency was estimated as described before.
The Chloramphenicol concentration in plates was 34 ug/ml for the high copy plasmids, 12.5 ug/ml for the medium/low copy plasmids and 12.5 for the three control strains transformed with the R6K plasmid.
BBa_K300000 construction: This section describes how this vector backbone was assembled using BioBrick parts.
- First step - <partinfo>BBa_K300982</partinfo> intermediate part construction:
- <partinfo>BBa_K300983</partinfo>, provided by Mr Gene DNA synthesis service (www.mrgene.com), was excised from its original shipping vector (pMK-RQ) through digestion with AvrII restriction enzyme (Roche). It was then isolated through a 1% agarose gel electrophoresis and gel-extracted (Macherey-Nagel NucleoSpin Extract II).
- <partinfo>BBa_K300008</partinfo> was assembled via BioBrick Standard Assembly from available parts and was excised from its vector (<partinfo>pSB1A2</partinfo>) through digestion with XbaI-SpeI (Roche). It was then isolated using the same procedure described above and it was dephosphorylated by using Antarctic Phosphatase (NEB).
- Digested <partinfo>BBa_K300983</partinfo> and <partinfo>BBa_K300008</partinfo>, all having compatible sticky ends, were ligated with T4 Ligase (Roche) and transformed into competent BW23474 E. coli strain (<partinfo>BBa_K300985</partinfo>) in order to allow the ligated plasmid propagation at high copy number. This strain was necessary because the replication origin of the resulting plasmid was the conditional R6K origin (<partinfo>BBa_J61001</partinfo>).
- Positive transformants, grown on Chloramphenicol (34 ug/ml) plates, were identified by restriction mapping with EcoRI-HindIII (Roche). The yielded plasmid was <partinfo>BBa_K300982</partinfo>.
- Second step - <partinfo>BBa_K300000</partinfo> final vector construction:
- <partinfo>BBa_K300982</partinfo> DNA was miniprepped, digested with EcoRI-PstI (Roche), run on agarose gel and gel-extracted as described above, in order to cut out and eliminate the RBS (<partinfo>BBa_B0033</partinfo>) between EcoRI and PstI.
- <partinfo>BBa_I52002</partinfo> DNA was excised from <partinfo>pSB4C5</partinfo> by EcoRI-PstI (Roche) digestion and isolated by gel run and gel extraction as described above.
- Digested <partinfo>BBa_K300982</partinfo> and <partinfo>BBa_I52002</partinfo> were ligated and transformed into competent DB3.1 E. coli strain (<partinfo>BBa_V1005</partinfo>), selected with Chloramphenicol at 34 ug/ml.
- Positive transformants were screened by restriction mapping with EcoRI-HindIII (Roche) and sequencing with VF2 (<partinfo>BBa_G00100</partinfo>) and VR (<partinfo>BBa_G00101</partinfo>) standard primers. The yielded plasmid was <partinfo>BBa_K300000</partinfo>.
All the DNA manipulations were performed according to manufacturer's protocols.
Integration protocol:
- Transform the <partinfo>BBa_J72008</partinfo> helper plasmid in the host strain (MC1061 or MG1655) and select transformants on Amp (50 ug/ml) plates under permissive conditions (30°C) overnight.
- Inoculate a single colony in selective LB and let the culture grow at 30°C, 220 rpm. When the culture reaches the OD600 of 0.4-0.6 prepare chemically competent cells.
- Transform the integrative vector with the desired insert in the BBa_J72008-containing strain and select co-transformants on Cm (34 ug/ml) plates under permissive conditions (30°C) overnight. At this temperature <partinfo>BBa_J72008</partinfo> can be replicated and so the pir protein product can be expressed in the cells. The pir product enables the propagation of the integrative vector by replicating the R6K origin.
- Inoculate a single colony in 5 ml of LB + Cm at 12.5 ug/ml and incubate the culture at 37°C, 220 rpm overnight. At this temperature the <partinfo>BBa_J72008</partinfo> helper cannot be replicated and the Phi80 integrase is expressed by the remaining copies of the helper. The bacteria that are able to grow in this selective medium should be correct integrants because the integrative vector cannot be replicated by the pir product anymore.
- Streak the culture on a Cm plate (at 12.5 ug/ml) and incubate it at 43°C overnight to ensure the loss of the helper plasmid. The bacteria that form colonies should be correct integrants without the <partinfo>BBa_J72008</partinfo> helper plasmid.
Validate the loss of the helper plasmid by inoculating colonies in Cm (at 12.5 ug/ml) media and counterselecting them in Amp (at 50 ug/ml) media. Validate the correct integration position by performing colony PCR with primers P1/P2, P3/P4, P1/P4, P2,P3 and VF2/VR. Validate the phenotype (when possible).
Expected amplicon length [bp] when the vector is integrated into the Phi80 locus:
|
| No integrant
| Single integrant
| Multiple tandem integrants (>1)
|
| VF2/VR
| none
| 280 + insert length
| 280 + insert length
|
| P1/P4
| 546
| 546 + insert length + 2171 (i.e. the BBa_K300000 length)
| 546 + insert length + 2171 (i.e. the BBa_K300000 length)
|
| P1/P2
| none
| 452
| 452
|
| P3/P4
| none
| 666
| 666
|
| P2/P3
| none
| none
| 572
|
Marker excision protocol:
- Inoculate an integrant in selective LB medium and let it grow to OD600=0.4-0.6. Prepare chemically competent cells.
- Transform the pCP20 helper plasmid in the competent strain and select transformants on Amp (100 ug/ml) plates under permissive conditions (30°C) overnight. At this temperature the pCP20 can be replicated. The pCP20 plasmid contains Amp and Cm resistance markers, a thermoinducible Flp recombinase expression system and a heat-sensitive replication origin. The permissive temperatures for the pCP20 propagation are the same as <partinfo>BBa_J72008</partinfo>.
- Inoculate a single colony in 5 ml of LB without antibiotic and incubate the culture at 37°C, 220 rpm overnight. At this temperature the pCP20 helper cannot be replicated and the Flp recombinase is expressed by the remaining copies of the helper. The bacteria that should loose the R6K origin and the Cm resistance upon FRT sites recombination, mediated by Flp.
- Streak the culture on a LB plate and incubate it at 43°C overnight to ensure the loss of the helper plasmid. The bacteria that form colonies should be without the pCP20 helper plasmid.
Validate the loss of the helper plasmid by inoculating colonies in Amp (at 100 ug/ml) media and validate the loss of the Cm resistance from the genome by inoculating colonies in Cm (at 12.5 ug/ml) media. Validate the correct length of the integrated part without Cm resistance and R6K origin by performing colony PCR with primers P1/P4 (which amplify the entire Phi80 locus) and VF2/VR (which amplify the integrated part). Validate the phenotype (when possible).
Colony PCR: a single colony or 1 ul of culture was added to the Invitrogen Platinum Taq reaction mix and was heated at 94°C for 10 min. Then it was assayed with this cycle (X 35): 94°C 30 sec, 60°C (for VF2/VR) or 63°C (for the other primers) 30 sec, 72°C according to the amplicon expected length (1Kb/min). Then the reaction was kept at 72°C for 10 min and it was run on a 1% agarose gel with the GeneRuler 1Kb Plus DNA ladder (Fermentas).
Fluorescence assays: integrants were inoculated in 1 ml of M9 + Cm (12.5 ug/ml) and grown at 37°C, 220 rpm overnight. The cultures were diluted 1:100 in 1 ml of selective M9 and let grow for about 4 hours under the same conditions as before. Three 200 ul aliquots for each culture were transferred to a 96-well microplate and assayed in the Infinite F200 microplate reader (Tecan) for about 20 hours with the following kinetic cycle: 37°C, 5 min sampling time, linear shaking 15 sec (amplitude=3), wait 5 sec, measure OD600, measure fluorescence with the proper filter (EX:nm/EM:540nm for GFP or EX:535nm/EM:620nm for RFP) with gain=50 or 70. The same protocol was followed for the MC1061 and the MG1655 non-integrant strains, which were grown in M9 without antibiotic.
Results
Validation of pir strains to propagate the R6K replication origin
The BW25141 (pir+) and BW23474 (pir-116) E. coli strains were chosen to propagate the vectors with the R6K replication origin at medium copy (~15 molecules per cell) and high copy (~250) respectively.
The results about their capability to propagate R6K plasmids are shown here:
| Strain
| Efficiency with no DNA
| Efficiency with pSB*** (positive control)
| Efficiency with the self-ligated <partinfo>BBa_K300008</partinfo> (R6K plasmid)
|
| BW25141 (<partinfo>BBa_K300084</partinfo>, pir+)
| 0
| 10^5
| 10^5
|
| BW23474 (<partinfo>BBa_K300085</partinfo>, pir-116)
| 0
| 10^6
| 10^6
|
| DH5alpha (<partinfo>BBa_V1001</partinfo>, neg control)
| 0
| 10^8
| 0
|
| MC1061 (<partinfo>BBa_K300078</partinfo>, neg control)
| 0
| 10^6
| 0
|
| MG1655 (<partinfo>BBa_V1000</partinfo>, neg control)
| 0
| 10^5
| six small colonies from 2 ng of DNA
|
These results show that the R6K conditional replication origin can be only propagated in pir+ and pir-116 strains (<partinfo>BBa_K300084</partinfo> and <partinfo>BBa_K300085</partinfo>), while the transformation of other strains with the R6K plasmid yielded no colonies after transformation. The exception was MG1655, for which six small unwanted colonies appeared on the selective plate. Plasmid DNA was purified/HindIII-digested/gel-run for at least one colony for BW25141-<partinfo>BBa_K300008</partinfo> and BW23474-<partinfo>BBa_K300008</partinfo>, while all the six colonies were analyzed for MG1655-<partinfo>BBa_K300008</partinfo> plate. The electrophoresis showed the expected length of the transformed DNA for all the clones except for the MG1655 six colonies, for which a smear was present in the lane (data not shown). Probably, Cm-resistant contaminants were present in the MG1655 culture during the preparation of competent cells.
For this reason, competent cells were prepared again for MG1655 and the transformation procedure was repeated for this strain, yielding no colonies in the <partinfo>BBa_K300008</partinfo> plate as expected.
Miniprep of the BW25141 and BW23474 strains transformed with <partinfo>BBa_K300008</partinfo> yielded a DNA concentration of ~20 ng/ul (qualitatively comparable with medium copy number plasmids) and ~90-100 ng/ul (qualitatively comparable with high copy number plasmids).
The results shown in the table above also show that the R6K plasmid in pir+ and pir-116 strains was transformed with the same efficiency as the pSB*** positive control plasmid, demonstrating that the R6K origin doesn't give any handicap in plasmid transformation.
So, the BW25141 and BW23474 strains can be successfully used to propagate the integrative vector after the excision of the pUC19-derived high copy replication origin, present in the default insert <partinfo>BBa_I52002</partinfo>.
Integration of the desired BioBrick part into the Phi80 genome locus
MC1061 and MG1655 were chosen as host strains for integration. <partinfo>BBa_K173001</partinfo> (constitutive strong promoter with GFPmut3) and the EcoRI-PstI fragment of <partinfo>BBa_J61002</partinfo>-<partinfo>BBa_J23101</partinfo> (here called PconRFP - constitutive strong promoter with RFP) were chosen as two proof of concept BioBrick parts to test the integration capability of the <partinfo>BBa_K300000</partinfo> vector in the Phi80 genome locus of these strains. For this reason, <partinfo>BBa_K173001</partinfo> and PconRFP were ligated in <partinfo>BBa_K300000</partinfo> (digested with EcoRI-PstI) and propagated using BW23474.
The integration protocol was performed as described in the Materials and Methods section for 4 different combination:
| Integrant name
| Strain
| Insert of <partinfo>BBa_K300000</partinfo>
|
| MC-GFP
| MC1061
| <partinfo>BBa_K173001</partinfo>
|
| MC-RFP
| MC1061
| PconRFP
|
| MG-GFP
| MG1655
| <partinfo>BBa_K173001</partinfo>
|
| MG-RFP
| MG1655
| PconRFP
|
Validation of the loss of BBa_J72008
Validation of the actual integration site
Validation of the integrants phenotype
Chloramphenicol resistance marker excision
Validation of the loss of pCP20 and the resistance marker
Validation of the marker-less phenotype
Discussion
A novel integrative vector for E. coli has been successfully designed, constructed and used to integrate two proof of concept protein expression systems in two commonly used E. coli strains.
The results showed that the vector is fully functional and can integrate into the correct targeted locus of the host chromosome through the Phi80 site-specific recombination system by using <partinfo>BBa_J72008</partinfo>, an existing BioBrick helper plasmid from the Registry. In most cases, the integration occurs in tandem copies, probably because of the too high Chloramphenicol concentration used during the selection of integrants, which forces multiple integration of Cm-resistant constructs. This concentration was the same used during the pSC101 low copy plasmid (~5 copies per cell) selection. In some cases, it is desirable to have a single copy of the desired BioBrick in the genome, for example when the gene dosage is important. In [Haldimann A and Wanner BL, 2001] the usage of Chloramphenicol at 6 ug/ml yielded a very high percentage of single integrants. However, when tested in our lab, the MG1655 strain could survive on LB plates with Cm at 6 ug/ml and also at 8 ug/ml. For this reason a higher concentration of Cm was chosen for selection. Further studies should investigate the optimal antibiotic concentration to yield the highest single integrants percentage as possible.
The Flp/FRT mediated excision of the R6K and, most importantly, of the Cm resistance marker also worked by using the pCP20 helper plasmid. The estimated efficiency of this process was 100%. In addition, multiple tandem integrants became single integrants after the marker excision. This is because the Flp recombinase mediated the recombination of all the FRT sites of the multiple integrants until only a single FRT site was present in the Phi80 locus. The marker excision is a powerful tool to engineer microbial strains for industrial protein manufacturing because the engineered organism should not carry unsafe antibiotic resistances that may be diffused in the environment.
The fluorescence phenotype confirmed the correct integration into the E. coli chromosome. As expected, in general multiple integrants showed a higher fluorescence than the single integrants.
The BioBrick compatibility and the vector modularity give the possibility to the scientific community to stably engineer novel biological functions in E. coli with a very easy and user friendly methodology. A user’s handbook about the vector usage is shared in the Registry, as well as the users experiences and the compatibility information.

Integrative standard vector for yeast

Self-cleaving affinity tags to easily purify proteins
 "
"











