 "
"
Team:NYMU-Taipei/Project/Speedy reporter
From 2010.igem.org
(→Design) |
(→aptamer) |
||
| Line 69: | Line 69: | ||
== [[Team:NYMU-Taipei/Project/Speedy reporter/Material and Methods| aptamer]] == | == [[Team:NYMU-Taipei/Project/Speedy reporter/Material and Methods| aptamer]] == | ||
| - | *We PCR the aptamer by ourselves based on sequence showed on paper (Valencia-Burton07). | + | *We performed PCR to get the needed aptamer by ourselves based on sequence showed on a paper of (Valencia-Burton07). |
| - | We first | + | We first designed primers that adding prefix in the front of the aptamer sequence and suffix in the end of the aptamer sequence. We then digested the aptamer with Xbal&PstI cutting enzymes and the pLac with Spal&PstI cutting enzymes. Then we then used ligase to join them together. |
==protein reporter device== | ==protein reporter device== | ||
Revision as of 13:47, 27 October 2010
| Home | Project Overview | Speedy reporter | Speedy switch | Speedy protein degrader | Experiments and Parts | Applications | F.A.Q | About Us |
Contents |
Overview design by figure
Abstract
- Our Speedy RNA+protein reporter effectively skips protein folding when reporting, thus reducing the time for a fluorescent response.
More specific insights into molecular mechanisms and gene regulation are essential for improvement in synthetic biology. Understanding these mechanisms requires time. Our speedy reporter for reporting RNA and protein expression in a cell effectively skips protein folding when reporting -- the most time cost period during gene expression, and thus reducing the time needed to get a fluorescent response. By speeding up the exploring for the rules of biological system in RNA and protein expression, we can not only help generate more novel circuits but also providing a tool for exploring gene regulation in the progress of synthetic biology.
Introduction
Recent studies of mRNA localization show that a great part of mRNA localize in specific cytoplasm position (Martin, 2009). For examples, ASH1 mRNA localize at bud tip of budding yeast to allow asymmetric segregation from mother to daughter cell (Paquin, 2007). In the Drosophila the localization of mRNA at anterior and posterior of oocyte play an important role in the developing embryo (Johnstone, 2001). Local translation of mRNAs in axonal growth cones helps axon navigate to it synaptic partners (Lin, 2007). β-actins mRNA localize at sites of active actins polymerization, cytoskeletal-mediate motility need mRNA translation (Huttelmaier, 2005). All the examples above is studies on eukaryotic system. there are a few studies of mRNA location in prokaryotic system. And most of synthetic biology designs on prokaryotic bacteria. The more basic rule of prokaryotic system we know, the more successful and speedy experiments we will have.
The common way to detect mRNA is RT-PCR, which can only be done in vitro but can’t in a real living cell. The common way to detect the protein is fusion a reporter protein such as GFP to report it, and the folding of GFP takes about four hours. In order to do both assay speedy and in vivo, we apply a novel technique Bimolecular Fluorescence Complementation, BiFC. In our design, we need not to wait four hours for folding of GFP to detect our protein fusion GFP. We can get our signal in few minute using this method. And it also can detect the mRNA both location and quantity (Demidov, 2006). We can use this method save about fours for protein assay and two hours for mRNA assay (Fig.1).
 Figure.1 compare to the traditional method of detecting mRNA and protein. our speedy reporter only need 3 min to obtain signal (Demidov, 2006). | BiFC is developed base on the technique Protein-fragment Complement Assay, PCA (Demidov, 2006; Barnard, 2008). Protein-protein interactions coupled to refolding of a pair of split enzymes in the PCA technique. The enzyme used in PCA has it activity only when two split parts reconstruct together. The activities of enzyme act as a detector of protein-protein interaction (Remy, 2007). While the BiFC technique use split fluorescent protein instead of split enzyme in the PCA. The split form of fluorescent protein alone has no fluorescence. Fluorescence appears when two split parts reassembly together immediately in few minutes. For mRNA detection, we design a system differ from BiFC’s protein-protein interaction to RNA-protein interaction. Where a GFP is split into two inactive parts and fused with two parts of the split-eIF4A protein, a kind of RNA binding protein. On the other hand, we designed an mRNA aptamer that the eIF4A protein can bind to. EGFP will fluoresce through the interaction of split eIF4A and its corresponding aptamer. Using this method, we can immediately detect mRNA quantity and location in vivo. For protein detection, we design another system of BiFC which RFP is splits into two inactive parts and fused with two parts of antibody light chain and heavy chain. And then we fused the antigen to target protein. When target protein fusion antigen appears, the light chain and heavy chain combine with antigen. And then split RFPs reconstruct and fluoresce. |
Design
- Our circuit design:


EGFP/ERFP + split eIF4A
- split GFP/RFP?
We split the EGFP/ERFP into two parts, a large N-terminal part and a small C-terminal part. The N-terminal part contains performed chromophore and has a very weak fluorescence that hard to detect. Only when it combines with the small C-terminal fragment, the fluorescence becomes very bright. And thus can detect the location of the target protein and mRNA. The split form of EGFP/ERFP has a very little chance to reconstruct while they freely suspense in the cytoplasm. So we seldom get the false-positive signal (Demidov, 2006).
- What is eIF4A?
eIF4A is an abbreviation of eukaryotic initiation factor 4A. It is a member of the DEAD-box RNA helicase protein family eIF4F (Zunakamu, 2002), and the DEAD-box is one of the largest subgroups of the RNA helicase protein family (Story, 2000). Eukaryotic translation initiation factor 4F (eIF4F) is a protein consist of eIF4A, eIF4E, and eIF4G. eIF4A is a helicase need ATP to unwind the secondary structure of mRNA untranslated region and make ribosome binds easier. eIF4E can binds to the cap structure of mRNA. eIF4G is like a scaffold of eIF4A and eIF4E helping them coordinate their functions. Without eIF4E and eIF4G the eIF4A alone exist much lower RNA helicase activity than complete eIF4F (Imataka et al., 1997).
eIF4A aptamer
- What is eIF4A aptamer? (Zunakamu, 2002)
The eIF4A aptamer generated has highly affinity for complete eIF4A and will combine the split eIF4A(as we described above) into a complete one. In the presence of eIF4A aptamer, it will inhibit the ATP hydrolysis and prevent the RNA substrate which is bind on the eIF4A to be unwind. It is proposed that the eIF4A structure is in a equilibrium between dumbbell-shaped and compact one in the solution. When in the presence of ATP and absence of RNA aptamer, the equilibrium will be shifted into the dumbbell-shaped eIF4A (Fig.2). When in an opposite condition, the equilibrium will be otherwise shifted into the compact one (Valencia-Burton, 2007).

- eIF4A aptamer Secondary structure:
- Structure predicted by RNAfold:
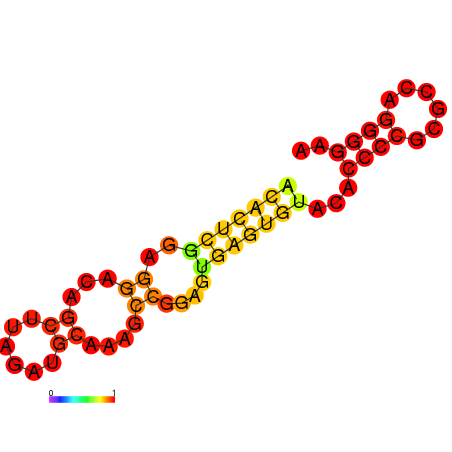 Figure.3 Aptamer Structure predicted by RNAfold
Figure.3 Aptamer Structure predicted by RNAfold - Structure of eIF4A aptamer:
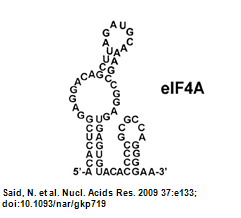 Figure.4 Aptamer Structure (Oguro, 2002)
Figure.4 Aptamer Structure (Oguro, 2002)
- Structure predicted by RNAfold:


Material and Methods
We constructed two devices by the parts below:
- RNA reporter consists of :
- EGFP
- ERFP
- eIF4A
- fusion parts
- aptamer
- protein reporter consists of :
- RFP
- split peptide adaptor
RNA reporter device
GFP
- Splitting the GFP(BBa_E0040) on the split point between 157th and 158th amino acid which was generated by iGEM07_Davidson_Missouri's BBa_I715019 and BBa_I715020.The A-part split is the same as BBa_I715019 ,but the B-part is one base different from BBa_I715020.After the spliting,we both add linkers in back of A-part split and B-part split via PCR.By doing so,we can get well-prepared for the next step.
RFP
- Splitting RFP (BBa_E1010) on the split point between 154th and 155th amino acid used by iGEM07_Davidson_Missouri's BBa_I715022 and BBa_I715023.The A-part split is the same as BBa_I715022, but the B-part has one base difference from BBa_I715023.After the spliting,we both add linkers in back of A-part split and B-part split via PCR.By doing so,we can get well-prepared for the next step.
eIF4A (need help)
- We take the protein coding region from the eIF4A mRNA transcript sequence from Mouse (from NCBI) and found it had 2 PstI cutting sites. For fear that our PstI cutting enzyme would cut the wrong place,we need to mutate the two PstI cutting sites.After mutation,we split eIF4A (BBa_K411100) at 215&216th amino acid.
- The template of eIF4A on a pGEX-4TI vector was kindly provided by Pro.C.Proud.
Fusion parts
GFP fusion system
- We fuse the split-GFP part with split-eIF4A part by PCR. So we get two sequences: split-GFP-A+linker+split-eIF4A-A(BBa_K41111)and split-GFP-B+linker+split-eIF4A-B((BBa_K11102))We add terminators in back of both sequences and insert them into one plasmid.
RFP fusion system
- We fuse the split-RFP part with split-eIF4A part by PCR. So we get two sequences:split-RFP-A+linker+split-eIF4A-A (BBa_K411103)and split-GFP-B+linker+split-eIF4A-B(BBa_K411104)
aptamer
- We performed PCR to get the needed aptamer by ourselves based on sequence showed on a paper of (Valencia-Burton07).
We first designed primers that adding prefix in the front of the aptamer sequence and suffix in the end of the aptamer sequence. We then digested the aptamer with Xbal&PstI cutting enzymes and the pLac with Spal&PstI cutting enzymes. Then we then used ligase to join them together.
protein reporter device
The basic principle of protein reporter device is the same as the RNA reporter. First, we fuse split RFP with anti-His tag antibody light chain and heavy chain. Second, we fuse His-tag sequence with our target protein sequence. Once our target protein sequence being tranlated the anti-His tag antibody will binding on the Histidine tag. And then with combining of the heavy chain and the light chain, The split ERFP reconstruct and make brightly fluorescence.

Advantage
1.Test the promotor strength in a speedy way.
- In traditional, the inducible promoter strength is test by the reporter gene (ex.GFP)behind the promoter. And need to wait for GFP folding for four hours. In our design, we can test promoter strength in the mRNA level and only need 3 min for GFP reconstruct.
2.Locate a specific gene or chemicals which can be heavy metals or so.
- Like the promoter testing, we can use the promoter that is heavy metal sensitive (ex. As or Zn). When the heavy metal is present the promoter will be active and transcript to mRNA which we can detect speedily.
3.Help to test other teams' biobricks.
4.Speed up the reporting progress.
- We can do a protein assay or mRNA assay in our speedy reporter system and get our signal in 3 mins. Faster than RT-PCR for mRNA need two hours and western blot for protein need 4.5 hours.
5.mRNA positioning in a sigle cell.
- The mRNA positioning in a bacteria help us know the synthetic biology rules in the bacteria.
6.Can measure the quantities of the mRNA.
7.View the temporal dynamics in a cell
References
- [Valencia-Burton07] Maria Valencia-Burton, Ron M McCullough, Charles R Cantor & Natalia E Broude. 2007. RNA visualization in live bacterial cells using fluorescent protein complementation. Nature method 4 (5), 421-427.
- [Oguro02] Akihiro Oguro, Takahashi Ohtsu, Yuri V. Svitkin, Nahum Sonenberg, and
- [Zunakamu02] Yoshika Zunakamu RA1 .2002. RNA aptamers to initiation factor 4A helicase hinder cap-dependent translation by blocking ATP hydrolysis.
- [Imataka97] Hiroaki Imataka, and Nahum Sonenberg. 1997. Human Eukaryotic Translation Initiation Factor 4G (eIF4G) Possesses Two Separate and Independent Binding Sites for eIF4A. Mol. Cell. Biol. 17:6940-6947
- [Story00] Randall M. Story, Hong Li*, and John N. Abelson. 2000. Crystal structure of a DEAD box protein from the hyperthermophile Methanococcus jannaschii.
- [Rackham04] Oliver Rackham and Chris M Brown. 2004. Visualization of RNA–protein interactions in living cells: FMRP and IMP1 interact on mRNAs.
- [Demidov06] Vadim V Demidov & Natalia E Broude,2006. Profluorescent protein fragments for fast bimolecular fluorescence complementation in vitro.
- [Llopis10] Paula Montero Llopis, Audrey F. Jackson, Oleksii Sliusarenko, Ivan Surovtsev, Jennifer Heinritz, Thierry Emonet, Christine Jacobs-Wagner.,2010. Spatial organization of the flow of genetic information in bacteria.
- [Muzzey09] Dale Muzzey, Alexander van Oudenaarden.,2009. Quantitative Time-Lapse Fluorescence Microscopy in Single Cells.
- [Lin07] Andrew C Lin and Christine E Holt,2007. Local translation and directional steering in axons. The EMBO journal 26:3729-3736
- [Huttelmaier05] Stefan Hüttelmaier, Daniel Zenklusen, Marcell Lederer, Jason Dictenberg, Mike Lorenz, XiuHua Meng, Gary J. Bassell, John Condeelis & Robert H. Singer., 2005. Spatial regulation of β-actin translation by Src-dependent phosphorylation of ZBP1. Nature 438:512-515
- [Martin09] Kelsey C. Martin, and Anne Ephrussi, 2009. mRNA Localization: Gene Expression in the Spatial Dimension. Cell 136:719-730
- [Paquin07] Nicolas Paquin and Pascal Chartrand,2007. Local regulation of mRNA translation: new insights from the bud. Trends in Cell Biology 18 , v3:105-111
- [Johnstone01] Oona Johnstone and Paul Lasko, 2001. TRANSLATIONAL REGULATION AND RNA LOCALIZATION IN DROSOPHILA OOCYTES AND EMBRYOS. Annu. Rev. Genet. 35, 365-406
- [Remy07] Ingrid Remy and Stephen W. Michnick, 2007. Application of protein-fragment complementation assays in cell biology. BioTechniques 42 (2),137-145
- [Barnard08] Emma Barnard, Neil V. McFerran, Alan Trudgett, John Nelson and David J. Timson. 2008. Development and implementation of split-GFP-based bimolecular fluorescence complementation (BiFC) assays in yeast. Biochem. Soc. Trans. 36, 479-482.