| August
|
| M | T | W | T | F | S | S
|
| | 1
|
| 2 | 3 | 4 | 5 | 6 | 7 | 8
|
| 9 | 10 | 11 | 12 | 13 | 14 | 15
|
| 16 | 17 | 18 | 19 | 20 | 21 | 22
|
| 23 | 24 | 25 | 26 | 27 | 28 | 29
|
| 30 | 31 |
-
|
|
|
|
October
01/10/2010
- Harvesting of the 50 plates (15 cm) from the previous day and picking of 50 clones were done, and the collected bacteria were incubated in a 700 mL LB culture for 3 hours with shaking, after which a megaprep (Invitrogen) was done.
- mini-preps of the 50 clones were done, and 10 samples were sent for sequencing.
02/10/2010
- Transfection of 13 T flasks (150 cm2, Hek 293 cells) was done, with the plasmid DNA purified the previous day (the shuffled cap genes library) and an adeno-helper plasmid, 35 micrograms from each plasmid for each flask, using HBSS for the transfection. A GFP control for transfection was also used.
- Analysis of the sequencing results for the 10 samples identified good shuffling of the cap genes. More mini-preps were inoculated for the same 50 clones.
03/10/2010
- Mini-preps for the samples from the previous day were done.
05/10/2010
- Harvesting of the cells for virus production was done by:
- Washing the flasks with the medium they contain, and collecting the media from 13 flasks in 500 ml corning conical centrifuge tubes.
- The cells were pelleted by centrifuging at 1500 rpm for 15 min at 4 ͦC
- Discard supernatant
- Washing: Resuspend the cells in 1X PBS and transfer to a 50 ml falcon
- Pellet the cells again by centrifuging at 1500 rpm for 15 min at 4 ͦC
- Discard supernatant
- Freeze-thaw cylces for cell rupturing:
- Add 20 ml lysis buffer to the cell pellet
- Put in liquid nitrogen for 5 min
- Thaw at 37ͦC
The cycles were repeated for 5 times, and the sample was frozen for next day.
06/10/2010
- The preparation for virus purification using iodixanol gradient was initiated according to the following:
- The sample from previous day was sonicated in a sonication bath for 1 min 20 sec.
- 50 units/ml benzonase were added to destroy RNA and DNA from non-AAV sources, which could later interfere with qPCR.
- The sample was incubated at 37 ͦC for 30 min, and vortexed every 10 min.
- The sample was then centrifuged for 15 min at 3270 xg
- The supernatant was transferred into a new 50 ml falcon
- Virus purification using iodixanol gradient centrifugation:
- A Pasteur pipette was plugged into a Beckman quick-seal centrifuge tube
- Using a 1000 µl pipette, 20 ml of the virus suspension were added the gradient was poured through the Pasteur pipette in the following order:
- 7 ml 15% Iodixanol solution
- 5 ml 25% Iodixanol solution
- 4 ml 40% Iodixanol solution
- 4 ml 60% Iodixanol solution
- The Pasteur pipette was carefully removed and the tube was sealed. A tare tube was prepared in the same way.
- Ultracentrifugation of the sample was carried out at 50,000 rpm for 2:30 hours at 4ͦC.
- The virus-containing phase is the 40% iodixanol phase, which was sucked out using a syringe and a needle. The AAV sample was divided into aliquotes, one of them stored at 4 ͦC for qPCR the next day, and the rest were frozen in liquid nitrogen then transferred to -20 ͦC.
07/10/2010
- qPCR of the sample from the previous day was carried out according to the protocol in the Methods section, and the titre of the viral library was determined to be 1.3*10^11 viral genomes/ml.
- The selection rounds of the viruses in the library were initiated on Huh-7 cells, the infection of cells was carried out as follows:
- AAV MOI 100, 10 and 1 were used on Huh-7 cells plated on 6-well plates, and the viruses were allowed to infect cells for 1 hour and 30 min.
- Adeno-virus 5 (MOI 10) was added then in a Biohazard II lab under the superviosion of lab personnel.
- Cells were incubated in the Biohazard II lab.
08/10/2010
- A non-ITR AAV helper vector was obtained and maxi-preped to clone into it the cap genes from the 50 single clones picked.
09/10/2010
- The non-ITR vector was cut with AscI and PacI, and so was the pTR-UF3 containg cap genes from the 50 clones. The bands were purified from the gel.
10/10/2010
- The cap genes were ligated into the non-ITR vector, and Hek 293 cells were prepared in 24-well plates for transfection.
- The first selection round Huh-7 cells infect with AAV library and Adeno-virus 5 were monitored under the microscope, and the typical cytopathic effect caused by Adeno-5 infection (roundening of cells) was observed. Cells were harvested by washing the wells with the media (in this case, one well of the 6-well plate was used).
11/10/2010
- The harvested cells from the previous day were incubated at 56 ͦC for 20 min to inactivate adeno virus 5. Then, freeze-thaw cycles (between liquid nitrogen and 37ͦC, see above) were carried out to lyse the cells and get the AAVs out. A 5-minute centrifugation at full speed was then done, and the supernatant containing the 1st selection round AAVs was collected and aliqueted and stored at -20ͦC (initial freezing with liquid nitrogen), or used immediately on the 2nd selection round Huh-7 cells as done previously. This time, instead of MOI for AAV, 100, 10 or 1 microliters of the supernatant from the first selection round were used on 3 wells of a 6-well plate, and the same volumes on the other 3 wells, however, for those the wells were washed and medium changed after 30 min of addition of AAV to increase the selection pressure. For both sets of wells, Adeno-5 (MOI 10) was added after 2 hours of initial addition of AAV.
- A triple transfection (1:1:1) of each non-ITR plasmid with cap genes from the 50 clones (see above) and Adeno-helper plasmid and a YFP construct with ITRs was carried out on Hek 293 cells in 24-well plates using FuGene. This was done to produce AAVs that contain YFP which will be expressed when these viruses successfully infect the target cells on which they will be tested. The viruses with best capsids will be able to infect target cells.
13/10/2010
- Mouse Primary Hepatocytes were received and plated on one 6-well plate plus two 24-well plates, which were previously coated with collagen.
- The 6-well plate with primary hepatocytes was infected with the library AAVs, after 1 hour and a half MOI 10 and 100 Adeno-5 were added, starting the first selection round on these cells
14/10/2010
- Viruses from the 24-well-plate-Hek 293 cells from two days before were collected after monitoring the expression of YFP in those cells. Freeze-thaw cycles were done as previously, and the supernatant after centrifugation was collected. Each well represented one AAV.
- 30 microliters of the supernatant collected for each AAV were used to infect target cells, which were:
- two 24-well plates HepG2 cells
- Two 24-well plates Huh-7 cells
- Two 24-well plates primary hepatocytes
15/10/2010
- AAVs from the second selection round on Huh-7 were collected from the supernatants after freeze-thaw cycles (see previous days), and stored at-20 C to infect the third selection round Huh-7 cells under the same conditions and in the same way as in the second selection round.
- AAVs from the first selection round on primary hepatocytes were harvested using freeze-thaw cycles as before (see previous days), and the supernatants were kept at -20 C for the second selection round (primary hepatocytes are received every Monday).
16/10/2010
- FACS measurements for the 6 24-well plates from two days earlier were done to measure YFP expression for the 48 different clones (AAVs!)in different kinds of cells. Cells were trypsinized (100 microliters trypsin) and then 500 microliters of buffer were added. Each well was measured for 1 min. The results showed no YFP expression for 47 of the samples in HepG2 and Huh-7, whereas one sample was positive (#43). This was confirmed further by microscopy. For the primary hepatocyes no sample showed any positive result.
Clone #43 was later injected into mice with a luc2-SV40 construct.
17/10/2010
- Huh-7 cells were infected with AAVs in the supernatant from the second selection round, thus starting the third selection round. The same conditions and volumes as in the second selection round were used.
18/10/2010
- The single sample of AAV that showed a positive result for YFP measurement was sent for sequencing.
- Primary hepatocytes were received and kept at 4 C for seeding the next day on 6-well plates. Collagen was used to coat the plates as before (Polymerization under UV for 1 hour was done).
19/10/2010
- Primary hepatocytes were seeded, and after 5 hours they were infected by AAVs from the first selection round. Adeno virus 5 MOI 100 was used, and varying volumes of the AAV-containing supernatant from the first selection round were applied.
21/10/2010
- Third selection round AAVs from Huh-7 and second selection round AAVs from primary hepatocytes were harvested by freeze-thaw cycles (see above).
- Viral DNA from the first, second and third selection rounds on Huh-7 and first and second selection rounds on primary hepatocytes was extracted using QIAmp MiniElute kit.
22/10/2010
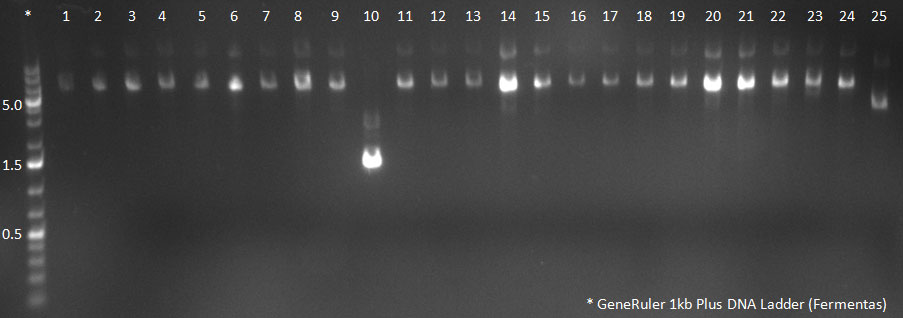
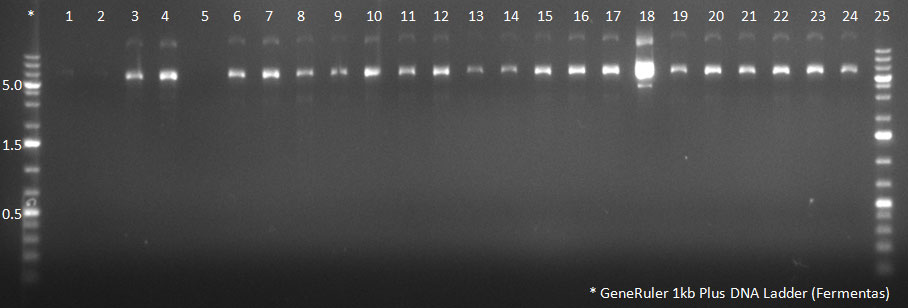
- A PCR reaction using a primer mix was carried out on the DNA extracted the previous day (plus 20 of the clones picked earlier and clones into a non-ITR vector, see above) to amplify the cap genes and introduce SwaI and SpeI sites, which were then used to clone the cap genes into a Wilson wild-type vector. There was a problem with the non-ITR vector used earlier, in the sense that the expression of Cap and Rep genes was not comparable, so not many functional viral particles could be produced, and for this reason the Wilson wildtype vector was used instead.
The conditions for the PCR were the same as the second PCR for amplification of the shuffled capsids (see above). The bands for cap genes (2.2 KB) were purified from the gel, then digested with SwaI and SpeI, together with Wilson wild-type vector (standard 20 microliter digestion in NEB buffer 4, for 2 hours at 25 C). Qiagen nucleotide removal followed for the cap genes, and the vector was gel-purified. A ligation reaction of cap genes into the vector was carried out with NEB Quick Ligase (according to the standard manufacturer's recommendations), for 1 hour 30 min at room temperature.
300px
PCR for Huh-7 clones
 PCR for Huh-7 clones
PCR for Huh-7 clones
 PCR for Huh-7 clones
PCR for Huh-7 clones
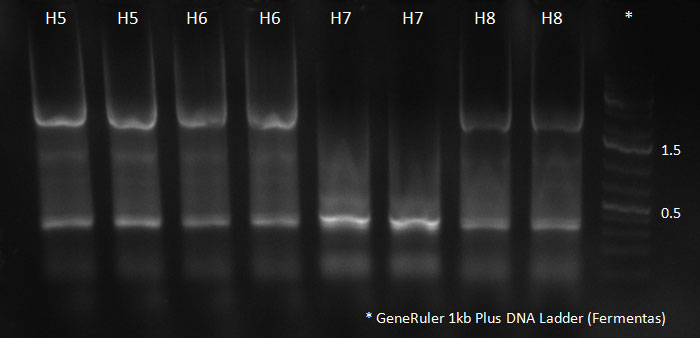
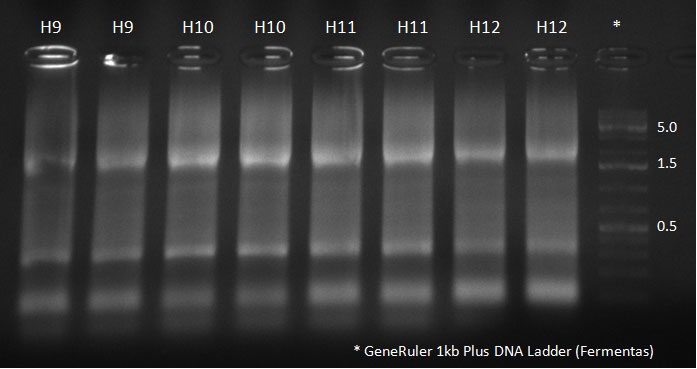
 Digested vector
Digested vector
23/10/2010
- The vector with cap genes from the previous day for the different selection rounds on Huh-7 and primary hepatocytes was transformed into Top10 cells.
24/10/2010
- 20 clones were picked from each selection round on Huh-7 and primary hepatocytes from the transformation on the previous day and used to inoculate mini-preps.
25/10/2010
- mini-preps were run for the samples from the previous day, and triple transfection of the Wilson construct with cap genes, Adeno-helper plasmid and a YFP construct with ITRs was done on 4 24-well plates of Hek293 cells in replica, each well containing an AAV construct from a different selection round on Huh-7 cells with different conditions during the selection process (washing after 30 min of AAV addition or no washing, see above).
From each selection round 12 samples were transfected, plus AAV wildtypes 1-12.
26/10/2010
- The AAVs were harvested from the cells of one set of the plates (see previous day) using freeze-thaw cycles, and 20 microliters of the supernatant after centrifugation were used to infect HeLa, Huh-7 and primary hepatocyte cells in a 96-well plate format. This was done in replicates. The plates are then used for FACS analysis of YFP expression.
- The clones picked were sent for sequencing.
27/10/2010
- A western blot and a dot blot for the other set of 24-well plates from two days earlier (see above) were initiated, with an antibody against a conserved region in cap genes.
 "
"







{kind=link}