Binding Site Design - September
13/09/2010
Restriction digest of psiCHECK-2 plasmid
This will be used as backbone for raPCR cloning. Enzymes: XhoI and NotI
Assay:
- 5 µL 10x NEBuffer 3
- 5 µL 10x BSA
- 5 µL plasmid (psiCHECK-2, ~370 ng/µL)
- 3 µL XhoI
- 1 µL NotI
- 18.6 µL H2O
Restriction digest was performed for approx. 5h
raPCR to create binding sites for different miRNAs
This random assembly PCR (raPCR) will be done to create binding site patterns for the miRNAs mentioned. In the first PCR step the oligos will basically anneal and constructs of different lengths will form. In the second step, the stop oligos are used as primers to amplify the previously formed constructs.
- first tries are: hsa-mir-122, hsa-mir122(ran9-12) and mm-mir-376a/375 (Oligos: ra001-003 and ra006)
stop-oligos: raPCR_AS13-stop_rev_NotI and raPCR_AS13-stop_fw_XhoI (ra017/018)
spacer: raPCR_AS13-spacer(0) and raPCR_AS13-spacer(10) (ra012/013)
Oligos were used in standard conc. (100µM)
| Oligo | mir-122 | mir-122(ran9-12) | mir-375/376a
|
| miR | 1 or 3 µL | 1 or 3 µL | 0.5 or 1.5 µL (each)
|
| spacer(0)or (10) | 1 µL | 1 µL | 1 µL
|
| stop | 0 or 0.5 µL each | 0 or 0.5 µL each | 0 or 0.5 µL each
|
Total: 12 reactions
each reaction was set up in 30 µL, using 2x Phusion Mastermix for 12 cycles
| Temp | Time |
|
| 95° | 05:00 |
|
| 95° | 00:30 | x 12 cycles
|
| 57° | 00:45
|
| 72° | 00:45
|
| 4° | forever |
|
- PCR purification: each PCR was purified using Qiagen PCR purification Kit and eluted in 32 µL
for the next PCR, three assay will tried:
- 5µL eluate + 1 µL of each stop oligo in 50µL total volume
- 5µL eluate + 2 µL of each stop oligo in 50µL total volume
- 20µL eluate + 1 µL of each stop oligo in 50µL total volume
stop-oligos: raPCR_AS13-stop_rev_NotI and raPCR_AS13-stop_fw_XhoI
In total there were 72 reactions. Each was run with 2x Phusion Mastermix, missing volume was filled with water.
| Temp | Time |
|
| 95° | 05:00 |
|
| 95° | 00:30 | x 25 cycles
|
| 65° | 00:45
|
| 72° | 00:50
|
| 72° | 05:00 |
|
| 4° | forever |
|
DNA was stored in fridge afterwards
14/09/2010
The 72 PCRs from 13/10/2010 were analysed on 1% agarose gel.

raPCR using a mixture of miRBS-375 and miRBS-376a oligos The initial 8 PCRs from the first raPCR were purified using Qiagen PCR Purification Kit, eluted in 32 µL and used as template for the second raPCR.
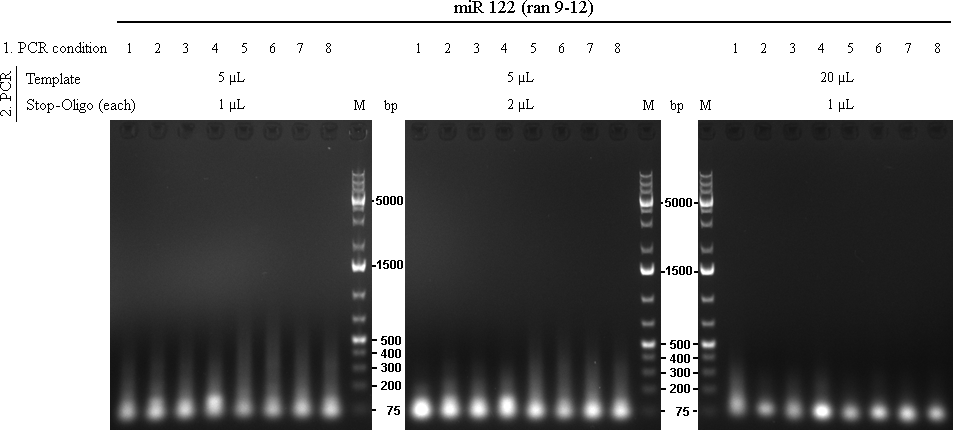
raPCR using miR-122 (ran9-12) oligos The initial 8 PCRs from the first raPCR were purified using Qiagen PCR Purification Kit, eluted in 32 µL and used as template for the second raPCR.
table 1: conditions for 1st raPCR
| 1. ra PCR condition | Template-Oligo [µL] | Stop-Oligo [µL] | Spacer-Oligo (1µL) [bp]
|
| 1 | 1 | 0 | 0
|
| 2 | 1 | 0 | 10
|
| 3 | 1 | 10 | 0
|
| 4 | 1 | 10 | 10
|
| 5 | 3 | 0 | 0
|
| 6 | 3 | 0 | 10
|
| 7 | 3 | 10 | 0
|
| 8 | 3 | 10 | 10
|
As we are looking for multiple binding sites, lanes with longest smear, meaning more long binding sites, were choosen for preparative gel:
Over all assay, using 5 µL template and 1µL of each stop-oligo seem to give the best result.
Following lanes were cut out of the gel for further cloning steps:
- Spacer(0): lane 5
- Spacer(10): lane 6
Samples for miR122 were applied to a preparative agarose gel (1.5%). Lanes were cut out from approx. 100 to 400bp and splitted at ~250bp. Therefore, we should have small binding site patterns (between 100 and 250bp) and larger binding site patterns (betweens 250 and 400bp) with either a shorter (Spacer(0)) or longer (Spacer(10)) spacer region.
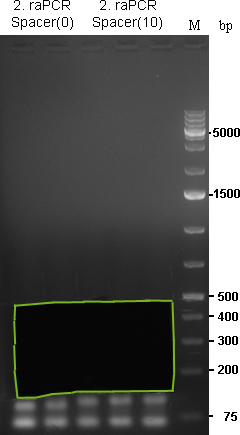 Preparative gel for 2. raPCR of miR-122 was cut out in the freamed region. |  Preparative gel for 2. raPCR of miR-122 (ran9-12) (1: Spacer(0), 2: Spacer(10)) and miR-275/376a (3: Spacer(0), 4: Spacer(10)) was cut out in the freamed region. |
As the gel volume was too much for dissolving in a single 2 mL tube, each part was splitted for dna extraction and brought together on the column. Gel extractions were done according to Qiagen Gel-extraction kit protocal and eluted in 30 µL water.
15/09/2010
Sample code:
- Spacer(0) - 100-250bp: 0S (for zero small)
- Spacer(0) - 250-400bp: 0L (for zero large)
- Spacer(10) - 100-250bp: 10S (for ten small)
- Spacer(10) - 250-400bp: 10L (for ten large)
- this System was used for each raPCR after gel purification
First row of cloning will be done with miR-122 samples. Others will follow.
Samples prepared: 122-0S,-0L,-10S,-10L
Gel extracted samples were digested with NotI/XhoI for cloning into psiCheck-2 vector:
5µL DNA (2µL for backbone) in a total volume of 30µL using 1µL XhoI and 0.6µL NotI enzyme, for 1.5 h at 37°C.
The digested DNA was then purified using Qiagen nucleotide removal kit and eluted in 30µL.
Subsequently, digested fragments were ligated over night at room temperature.
Ligation assay for Fermentas T4 ligase:
2µL Buffer
1µL Ligase
7µL water
1µL Backbone (6000bp, psiCHECK-2)
9µL purified restriction digest
16/09/2010
Transformation of ligations:
5µL ligation assay in 50µL TOP10 E.coli
25 min on ice
45sec heat shock on 42°C
1.5-2h shaking at 37°C
plated 200µL on Ampicillin-LB/Agar-Plates
after incubating ~8h, at 37°C, the plates were incubated overnight at room temperature
17/09/2010
Colonies were visible in reasonable numbers on every plate
Colony PCRs were performed to check for positive clones.
Primer for colony PCR were stop-oligos, used in the raPCR. The PCR was performed in a total volume of 20 µL.
One colony was dissolved in 20µL water. 5µL of this bacteria solution was used as PCR template. PCR conditions as recommended from Fermentas (see link above).
From each plate, 10 colonies were picked (40 in total).
Colony PCRs were then analysed on 1.5% agarose gel. Result: all negative
Troubleshooting....
Minipreps were prepared (5mL - LB-ampicillin) for each sample for text digestion (over night, shaking @37°C)
18/09/2010
Plasmid DNA was purified from over night cultures using Qiagen Plasmid Miniprep Kit according to the protocol. Elution was performed in 30 µL water.
Concentrations ranged from approx. 400 to 788 ng/µL.
Test digestion with NotI/XhoI was performed for 1h at @37°C and analysed on an 1.5% agarose gel. No insert was visible.
19/09/2010
Troubleshooting: Possible Problems
- we got the wrong backbone (given by Stefan M.) -> assure we have the right one
- backbone was not fully digested -> several test digestions
- insert was not fully digested -> can not be checked
- enzymes out of function? -> single digest of the vector can check that
- ligation did not work -> reaction was performed according to usual lab routine and protocol
- low transformation efficiency -> reaction was performed according to usual lab routine and protocol
- bacteria are not competent -> they work for other transformations
Testing steps:
- digestion of psiCHECK-2 given by Stefan M.
- test-digestion of psiCHECK-2 given by Stefan M. again and compare both
- repeat all steps
Over night digestion of backbone was performed at 37°C. 0.5 µg DNA was digested with 1 µL Enzyme in NEB Buffer 3 + BSA in a total volume of 30µL
20/09/2010
Digestion of psiCHECK-2 plasmid was analysed on 1% agarose gel:
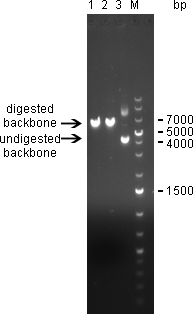
Single digest of psiCHECK-2 Plasmid was digestet overnight @37°C with NotI/XhoI using NEB Buffer 3 + BSA in a total volume of 30µL. 1)Digest with NotI 2)Digest with XhoI 3)undigested plasmid
Here we can see that both enzymes cut the plasmid. The linearized vector (visible at 6 kpb) in general shows up as a higher band than its undigested version, which is here visible at 4 kbp. Where the undigested plasmid shows concatemers, those are not visible after digestion, which proofs again for succesful digestion.
The new digested plasmid-backbone was used for repeat of the ligation.
21/09/2010
Ligation was performed for 4h @25°C (1µL NotI/XhoI-linearised psiCHECK2-plasmid + 4µL digested raPCR product) using Fermentas T4 Ligase
28/09/2010
- send 1.3, 1.5, 1.7, 3.8, 3.2 for sequencing
28/09/2010
- send 1.3, 1.5, 1.7, 3.8, 3.2 for sequencing
29/09/2010
- got sequencing results of 1.3, 1.5, 1.7, 2.8, 3.2.
- 1.3 - 3 binding sites - all ok
- 1.5 - 2 binding sites - both ok
- 1.7 - 3 binding sites - all ok
- 2.8 - not ok
(all those were made with Spacer(0) )
- 3.7 - 2 binding sites, created by Spacer(10) - both ok seperated by 10 nucleotide spacer
30/09/2010
- raPCRfrom above(1, 2, 3, 4)
- PCR purification ( nanodrop: c ˜ 100ng/µl)
- digested: 2 x 1 µg DNA:
-
- gel purification (nanodrop: c ˜ 25ng/µl)
- Ligation (Quick Ligase and overnight ligation with T4 ligase) into pSB1C3
- Vector ˜ 2000 bp
- Insert ˜ 200 bp
 "
"


_fertig.png)



{kind=link}